Page 16 of 26
PA31.{5,7} | Diabetes Mellitus & Pancreatic Cancer — SDL Guide (Part 2)
Type 2 DM — Insulin Resistance and β-Cell Failure
T2DM is a disease of progressive metabolic failure with two core defects that are present simultaneously but whose relative contribution shifts over time: insulin resistance and β-cell dysfunction.
Step 1 — Insulin resistance (the initiating event):
- Defined as an impaired biological response to normal circulating insulin concentrations
- Principal sites: skeletal muscle (reduced glucose uptake via GLUT4), liver (continued gluconeogenesis despite high insulin = hepatic insulin resistance), and adipose tissue
- Obesity — particularly visceral/central adiposity — is the dominant modifiable driver:
- Visceral fat releases free fatty acids (FFAs) and pro-inflammatory adipokines (TNF-α, IL-6, resistin) that interfere with insulin receptor signalling
- Adiponectin (insulin-sensitising adipokine) is paradoxically reduced in obesity
- Lipid accumulation in muscle and liver cells impairs the insulin receptor substrate-1 (IRS-1) → phosphatidylinositol-3-kinase (PI3K) → GLUT4 translocation pathway
- Metabolic syndrome — the cluster of insulin resistance, central obesity (waist circumference >90 cm in Asian males, >80 cm in Asian females), hypertriglyceridaemia, low HDL, hypertension, and impaired fasting glucose — is the key context for T2DM
Step 2 — β-cell compensation (early, maintained):
- In the face of insulin resistance, pancreatic β cells increase their output of insulin (hyperinsulinaemia)
- Blood glucose remains normal — this is the pre-diabetes phase (impaired fasting glucose 5.6–6.9 mmol/L; impaired glucose tolerance)
Step 3 — β-cell failure (progressive, irreversible):
- Chronic overstimulation, glucotoxicity, lipotoxicity, and oxidative stress lead to β-cell apoptosis and reduced β-cell mass
- Islet amyloid deposition (from IAPP — islet amyloid polypeptide, co-secreted with insulin) accelerates β-cell loss in T2DM
- Eventually, even maximum β-cell output is insufficient → overt hyperglycaemia → T2DM diagnosis
- Over decades, β-cell function continues to decline → ~30% of T2DM patients eventually require insulin
Genetic predisposition: Heritability ~50–70%. Key susceptibility genes: TCF7L2 (strongest effect), PPARG, KCNJ11 (Kir6.2 — the ATP-sensitive K⁺ channel subunit, same as the sulfonylurea target). These are polygenic, unlike the monogenic MODY mutations.
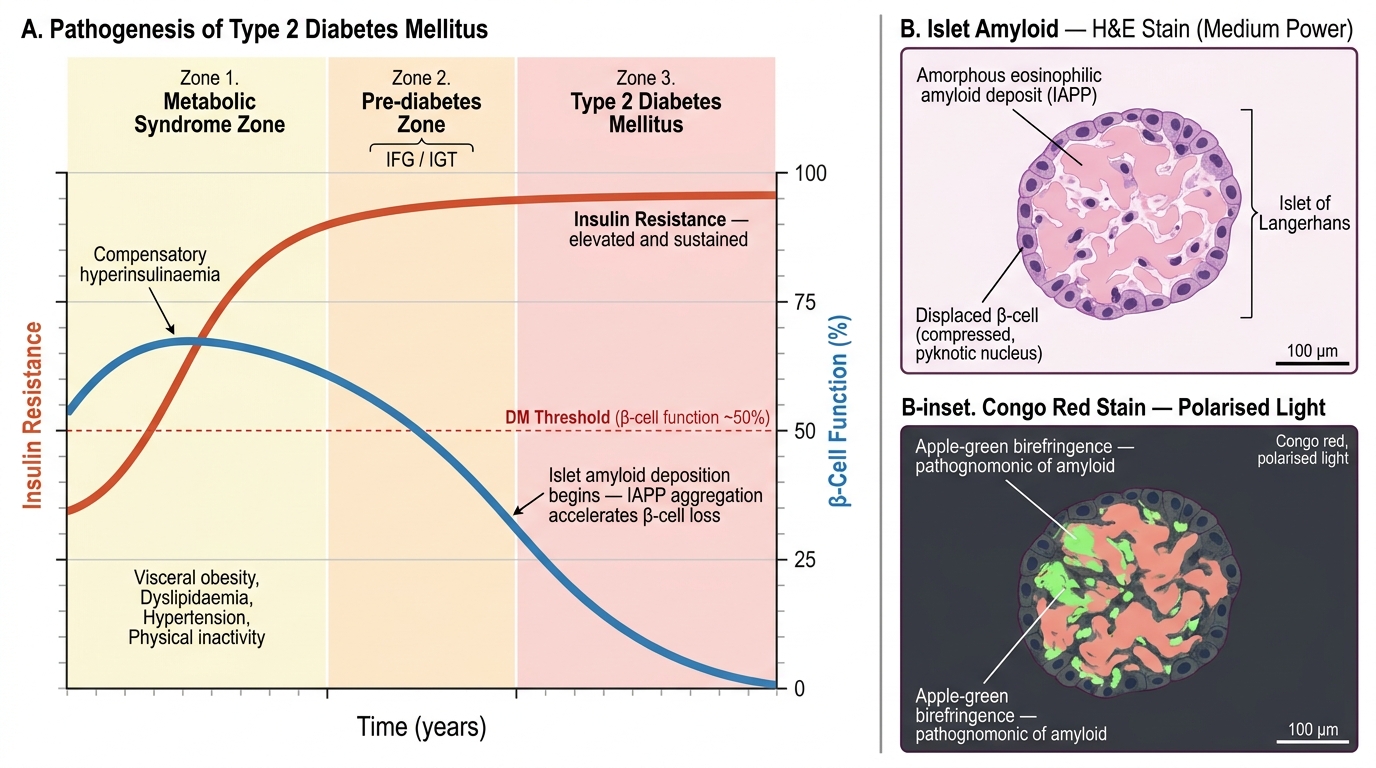
Pathogenesis of Type 2 Diabetes Mellitus: Natural History Timeline and Islet Amyloid Histology
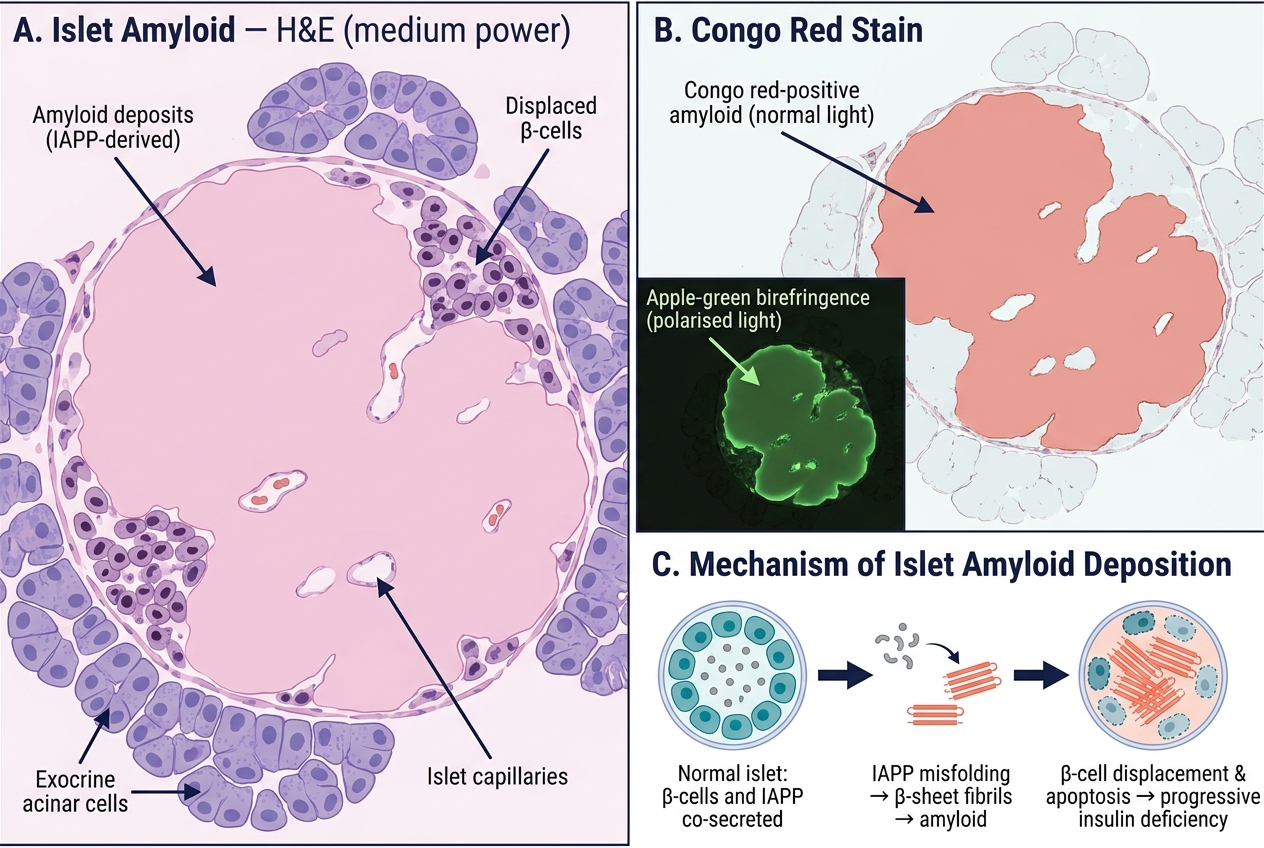
Islet Amyloid in Type 2 DM: H&E Histology, Congo Red Birefringence, and IAPP Deposition Mechanism
Diagnostic Laboratory Criteria for Diabetes
The ADA 2024 diagnostic criteria for diabetes mellitus are based on four tests, any one of which (confirmed on a second occasion if asymptomatic) is sufficient for diagnosis:
| Test | Diabetes threshold | Pre-diabetes range | Notes |
|---|---|---|---|
| Fasting plasma glucose (FPG) | ≥ 7.0 mmol/L (126 mg/dL) | 5.6–6.9 mmol/L (100–125 mg/dL) — IFG | Fasting = no caloric intake for ≥8 hours |
| 2-hour plasma glucose (2hPG) in OGTT | ≥ 11.1 mmol/L (200 mg/dL) | 7.8–11.0 mmol/L (140–199 mg/dL) — IGT | 75 g oral glucose load; used in GDM screening |
| HbA1c | ≥ 48 mmol/mol (≥ 6.5%) | 39–47 mmol/mol (5.7–6.4%) | Reflects mean glucose over 8–12 weeks; NOT valid in haemoglobinopathies, haemolytic anaemia, pregnancy |
| Random (casual) plasma glucose | ≥ 11.1 mmol/L (200 mg/dL) + symptoms | — | Classic symptoms: polyuria, polydipsia, unexplained weight loss |
HbA1c — the biochemistry:
- Glycosylated haemoglobin formed by non-enzymatic attachment of glucose to the N-terminal valine of the β-globin chain of HbA
- Reflects time-weighted average blood glucose over the lifespan of the red cell (~120 days)
- Result reported as % (NGSP/DCCT) or mmol/mol (IFCC); 6.5% ≈ 48 mmol/mol
- Conditions that falsely lower HbA1c: haemolytic anaemia, iron-deficiency anaemia with erythroid hyperplasia, recent blood transfusion, HbS/HbC — use FPG or OGTT in these patients
- Conditions that falsely raise HbA1c: iron-deficiency anaemia (reduced RBC turnover), splenectomy, renal failure
OGTT (oral glucose tolerance test):
- Patient fasts overnight → baseline plasma glucose → drinks 75 g glucose in 250 mL water over 5 minutes → plasma glucose at 2 hours
- Used when FPG is borderline (6.1–6.9 mmol/L) or for GDM screening (modified 50 g challenge at 24–28 weeks, then 100 g diagnostic OGTT if positive)
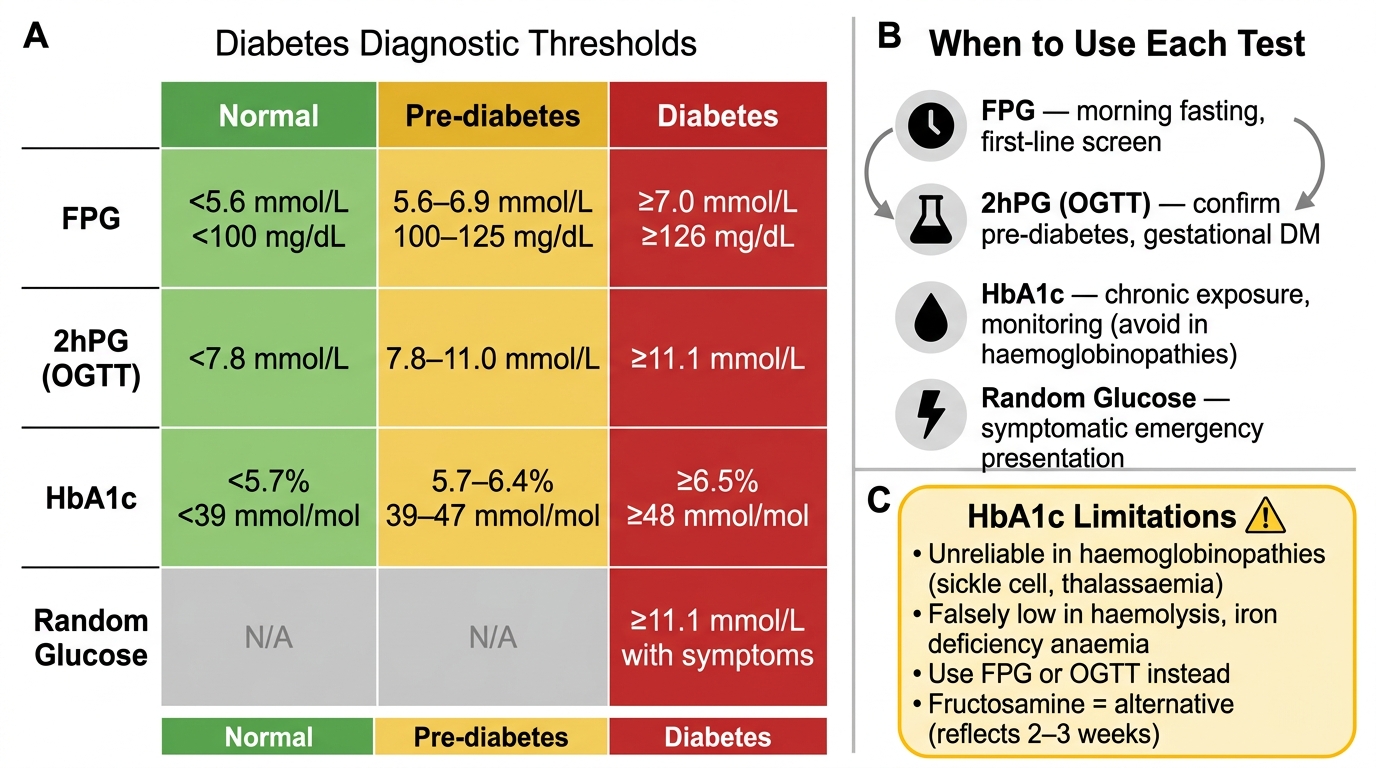
Diabetes Diagnostic Thresholds — Four Tests, Three Zones
SELF-CHECK
A 38-year-old woman with known sickle cell trait (HbAS) has been consistently symptomatic with polydipsia and polyuria for 3 months. Her HbA1c is 5.9% (41 mmol/mol). Which test should you order to confirm or exclude diabetes?
A. Repeat HbA1c in 3 months
B. Fasting plasma glucose or OGTT
C. Urine microalbumin
D. Fructosamine assay only if OGTT is inconclusive
Reveal Answer
Answer: B. Fasting plasma glucose or OGTT
HbA1c is unreliable in haemoglobinopathies (including sickle cell trait) because abnormal haemoglobins have altered red cell survival and can interfere with the assay. Fasting plasma glucose or OGTT are the correct diagnostic tests in this setting. Fructosamine (reflects 2–3 weeks of glycaemic control) is an alternative when HbA1c is invalid, but FPG/OGTT are first-line. Urine microalbumin screens for established nephropathy — it does not diagnose DM.
Acute Complications — DKA vs Hyperosmolar Hyperglycaemic State
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycaemic state (HHS) are the two acute life-threatening hyperglycaemic emergencies. Their differing pathophysiology reflects the underlying degree of insulin deficiency.
DKA — absolute insulin deficiency (classic in T1DM, rare in T2DM):
Pathogenesis:
1. Absolute insulin deficiency → no glucose uptake in insulin-dependent tissues
2. Counter-regulatory hormones rise (glucagon, cortisol, catecholamines) → gluconeogenesis and glycogenolysis → severe hyperglycaemia
3. Adipocyte lipolysis → massive free fatty acid (FFA) release → liver β-oxidation → ketone body synthesis (acetoacetate, β-hydroxybutyrate, acetone)
4. Ketone bodies accumulate → metabolic acidosis (anion gap) + acetone exhalation → fruity/ketotic breath
5. Osmotic diuresis → profound volume depletion + total body K⁺ depletion (despite hyperkalaemia on initial labs — K⁺ shifts out of cells in acidosis)
Diagnostic triad: hyperglycaemia (>11 mmol/L) + ketonuria/ketonaemia + metabolic acidosis (pH <7.3, bicarbonate <15 mEq/L)
Kussmaul respiration (deep, sighing breathing): a physiological compensation to expel CO₂ and correct acidosis
HHS — severe relative insulin deficiency (predominantly T2DM in elderly):
Pathogenesis:
1. Severe insulin deficiency but some residual insulin — enough to suppress lipolysis → no ketosis
2. Extreme hyperglycaemia (>33.3 mmol/L, often 55–100 mmol/L)
3. Massive osmotic diuresis over days to weeks in a patient who cannot compensate with fluid intake (elderly, institutionalised)
4. Serum osmolality >320 mOsm/kg → mental status changes, seizures, coma
5. No acidosis (pH normal, no ketonuria)
DKA vs HHS comparison:
| Feature | DKA | HHS |
|---|---|---|
| Type of DM | T1DM (mainly) | T2DM (mainly) |
| Glucose | >11 mmol/L (often 15–30) | >33.3 mmol/L (often 55+) |
| Ketones | +++ | Absent or trace |
| pH | < 7.3 | Normal (≥7.3) |
| Bicarbonate | < 15 mEq/L | Normal |
| Serum osmolality | Mildly elevated | > 320 mOsm/kg |
| Mental status | Variable | Often obtunded/coma |
| Mortality | 1–5% | 5–20% |
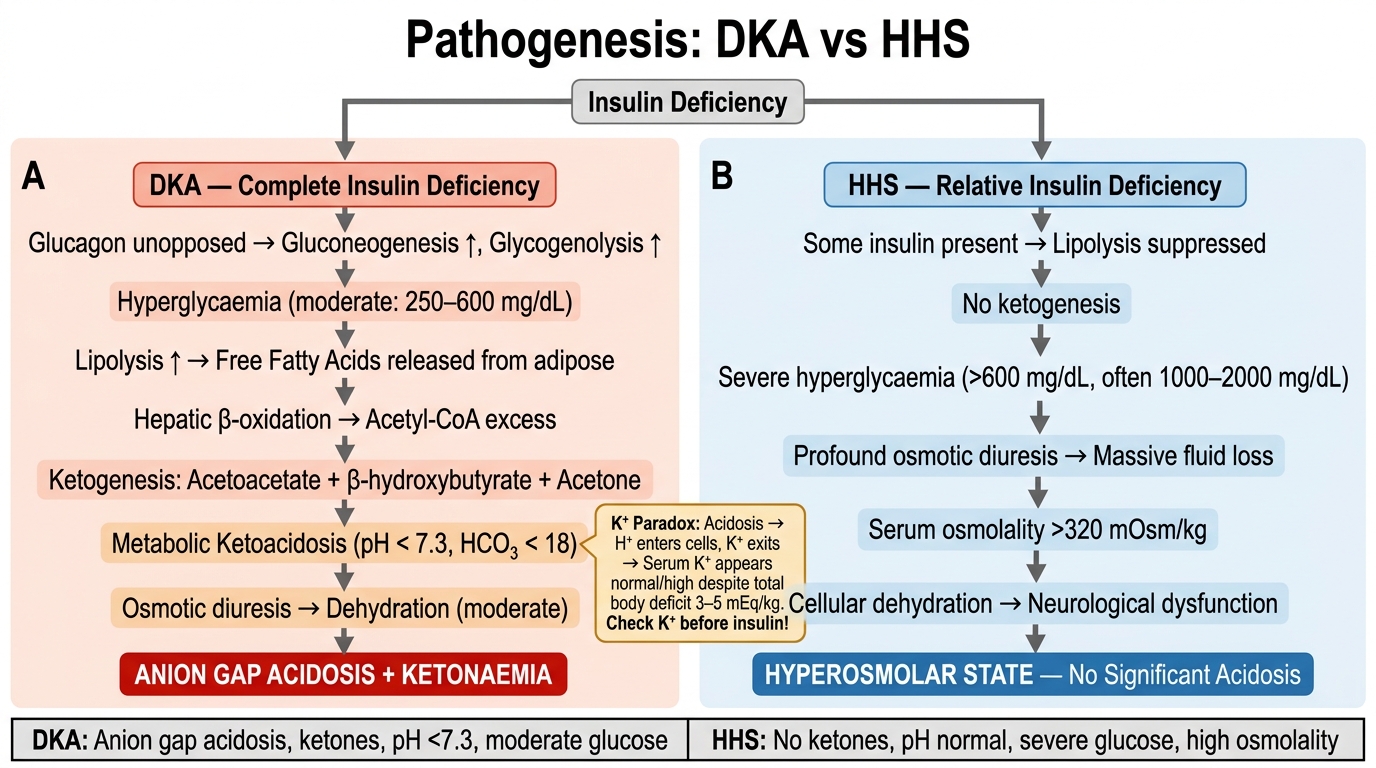
DKA vs HHS: Parallel Pathogenesis Flowchart
CLINICAL PEARL
The Potassium Paradox in DKA: On the initial blood gas and electrolytes, DKA patients are often hyperkalaemic despite a total body potassium deficit of 3–5 mEq/kg. This is because acidosis drives K⁺ out of cells (H⁺ in / K⁺ out exchange). However, once insulin is given and the acidosis corrects, K⁺ shifts back into cells rapidly — serum K⁺ can plummet to dangerous levels (cardiac arrhythmia, paralysis). Always check potassium before starting insulin in DKA; if K⁺ < 3.5 mEq/L, replace potassium first, then start insulin. This is a classic 'kill by treating' scenario that exams test repeatedly.