Page 5 of 24
PA2.4-5 | Cell Death — Necrosis, Apoptosis, Gangrene & Calcification — SDL Guide
Learning Objectives
- Define necrosis and describe its nuclear and cytoplasmic morphological hallmarks
- Classify the five major types of necrosis and relate each to its clinical context
- Define apoptosis and distinguish its morphology from necrosis
- Outline the intrinsic (mitochondrial/BCL-2) and extrinsic (death-receptor/Fas) apoptotic pathways
- Contrast necrosis and apoptosis across pathogenesis, morphology, and inflammation
- Define gangrene and differentiate dry, wet, and gas gangrene
- Distinguish dystrophic from metastatic calcification by mechanism, serum calcium, and clinical setting
INSTRUCTIONS
Cell death is the final common pathway of almost every disease process you will encounter in clinical practice. Understanding how and why cells die — and the morphological footprints they leave behind — underpins histopathology, clinical diagnosis, and treatment rationale. This module bridges Year-1 cell biology with Year-2 systemic pathology: you will recognise necrosis on slides, trace apoptosis signals to caspase activation, and explain calcification in surgical specimens. Robbins Basic Pathology, Chapter 2 (Cell Injury, Cell Death, and Adaptations) is the primary reading.
References
- Robbins & Kumar: Basic Pathology, 11th ed., Ch 2 (textbook)
- Harsh Mohan: Textbook of Pathology, 8th ed., Ch 2 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 60-year-old man is brought in unconscious after a 6-hour history of severe chest pain. His ECG shows an anterior STEMI. Emergency angioplasty restores flow to the left anterior descending artery, but the echo next morning reveals a 4 cm zone of pale, firm, non-contractile myocardium. Three months later, a routine biopsy of a cervical lymph node in another patient shows a granuloma with a chalk-white centre — calcium deposits visible to the naked eye.
Two patients, two very different cell death stories. By the end of this module you will be able to name exactly what killed each set of cells, explain the morphological changes on a microscope slide, and predict what the calcium in the lymph node means for serum biochemistry.
WHY THIS MATTERS
Cell death patterns are tested in every major exam — Theory, Practical spotters, and MBBS PG entrance. Recognising necrosis subtypes on H&E slides is a Year-2 core competency (PA2.4). Gangrene and calcification are surgical emergencies and frequent short-answer questions (PA2.5). Clinically: knowing whether a patient's tissue died by necrosis or apoptosis determines whether inflammation is fuelling ongoing organ damage — a principle that drives treatment in sepsis, autoimmunity, and chemotherapy.
RECALL
Before reading on, briefly recall from Year-1:
• What is the role of mitochondria in ATP generation?
• What happens to cell volume when Na⁺/K⁺-ATPase fails?
• What is the difference between intracellular and extracellular calcium concentration?
• What is a lysosome and which enzymes does it contain?
If any of these feel shaky, spend 5 minutes revisiting PY cell membrane transport — it will make the necrosis mechanisms click much faster.
Necrosis: Definition and Morphological Hallmarks
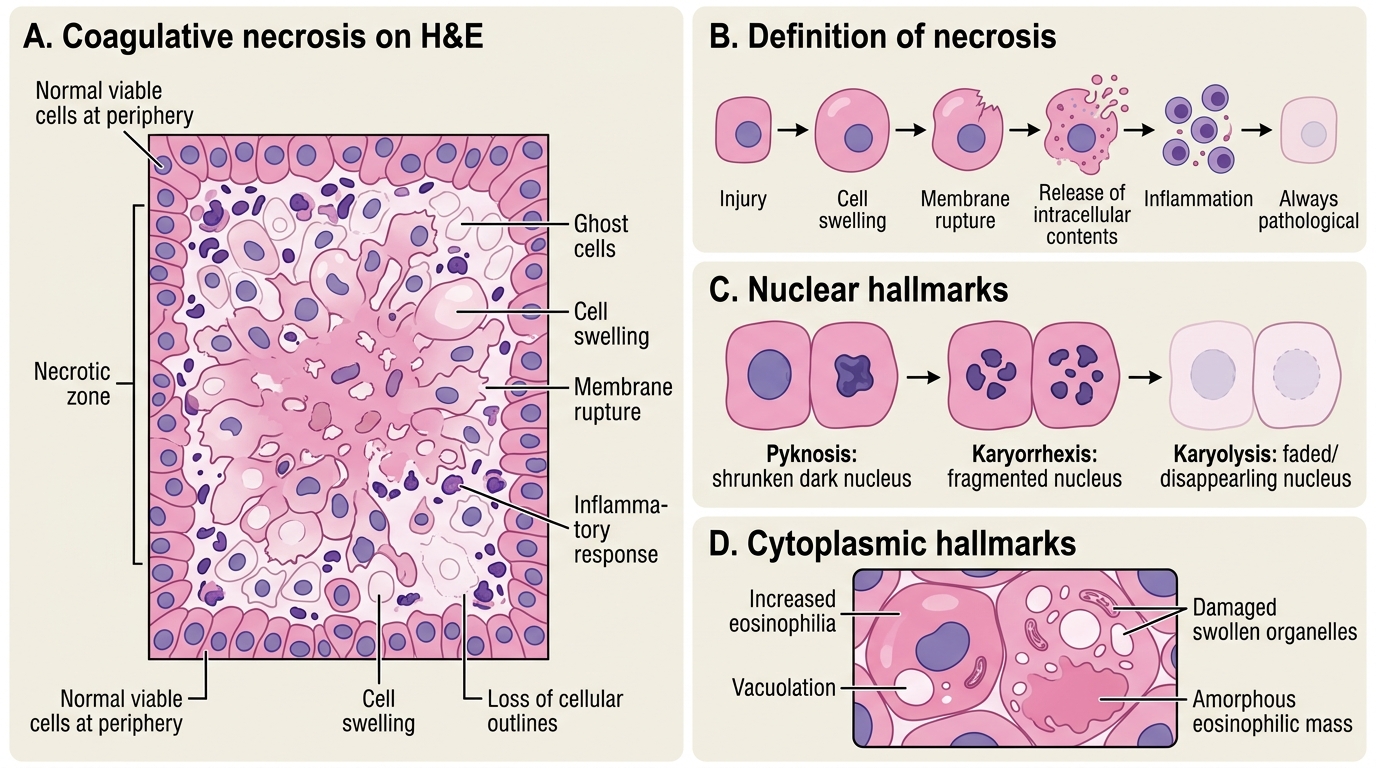
Necrosis: Morphological Hallmarks on H&E
Necrosis is a form of cell death characterised by cell swelling, membrane rupture, and release of intracellular contents that triggers an inflammatory response. It is always pathological — the cell does not orchestrate its own death.
Nuclear changes (the most diagnostically useful markers on H&E):
• Pyknosis — nucleus shrinks and becomes deeply basophilic (dark, condensed chromatin). The earliest nuclear sign.
• Karyorrhexis — pyknotic nucleus fragments into irregular basophilic clumps, scattered through the cytoplasm.
• Karyolysis — nucleus fades and disappears as DNases digest chromatin, leaving a ghost cell outline.
Cytoplasmic changes:
• Increased eosinophilia (pink staining) — denatured proteins bind eosin more avidly; ribosomes are lost (ribosomes normally contribute cytoplasmic basophilia).
• Vacuolation from swollen, damaged organelles.
• Loss of cellular outlines in advanced necrosis — cells merge into an amorphous eosinophilic mass.
IMPORTANT: Nuclear changes (pyknosis → karyorrhexis → karyolysis) are sequential but not always all visible in the same field. In an exam spotter, spotting even one of the three confirms necrosis.
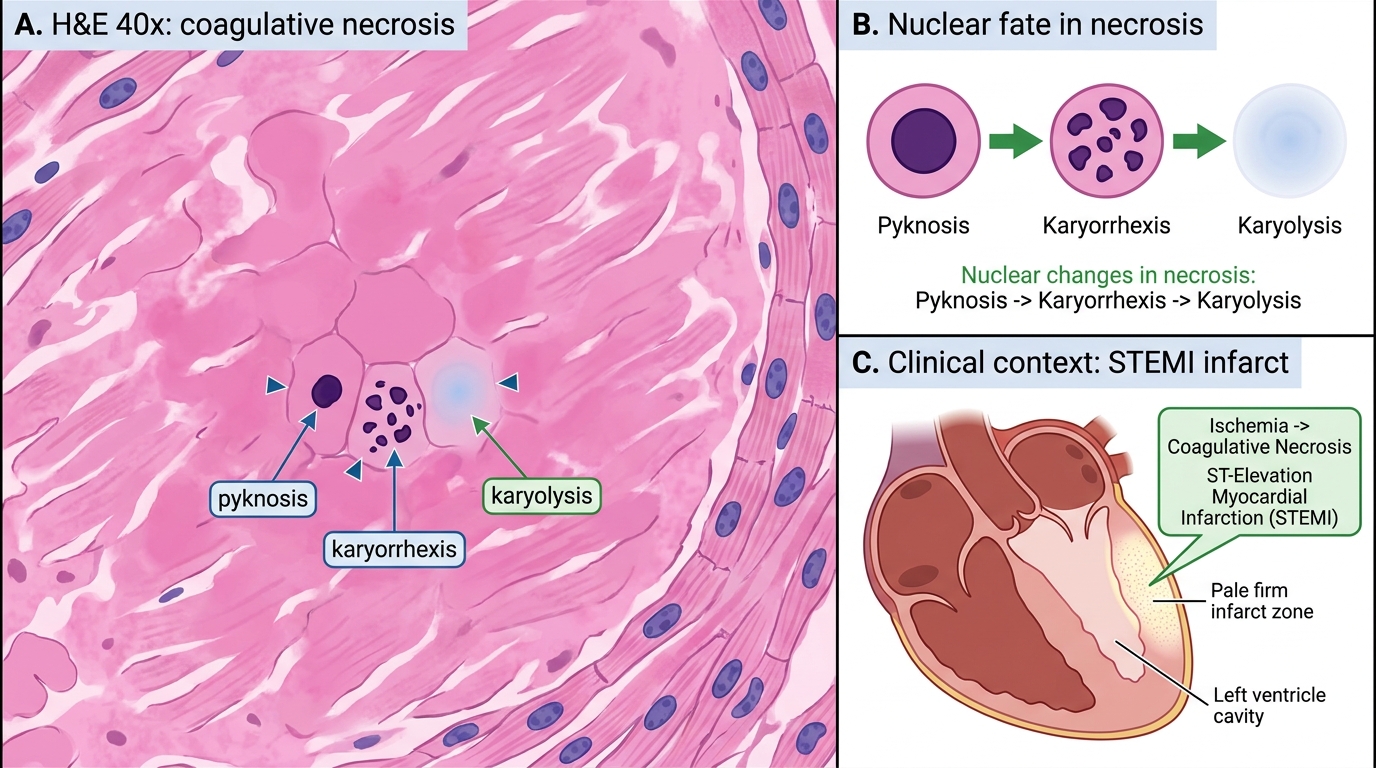
Nuclear Changes in Coagulative Necrosis
Types of Necrosis
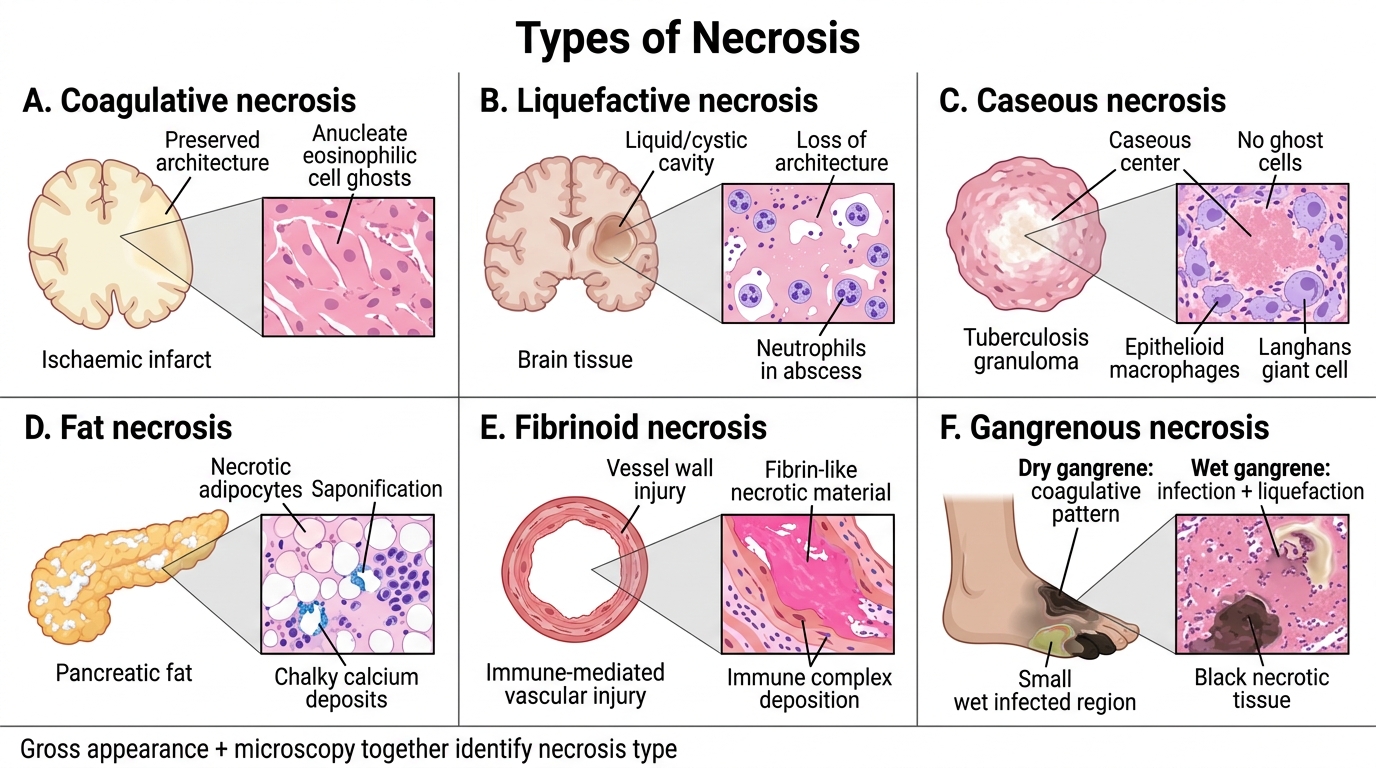
Types of Necrosis
The gross and microscopic appearance of necrosis differs by organ and insult. Six types are high-yield.
1. Coagulative necrosis
Prototype: myocardial infarction, renal infarct, splenic infarct.
Mechanism: ischaemia denatures both structural proteins and lysosomal enzymes simultaneously, so the cell ghost (cellular outlines) is preserved for days to weeks — architecture is recognisable even though cells are dead.
Gross: pale, firm, yellow-white zone (early infarct); later, softened and yellow.
Microscopy: anucleate, eosinophilic cell ghosts with preserved architecture.
2. Liquefactive necrosis
Prototype: cerebral infarct, pyogenic (bacterial/fungal) abscess.
Mechanism: abundant lysosomal enzymes (brain is rich in lipases; neutrophils in abscess secrete elastases, proteases) digest all proteins → liquid, creamy, or cystic cavity.
Gross: soft, liquid, cystic space (brain); pus-filled cavity (abscess).
Microscopy: loss of all architecture; fluid or cellular debris with neutrophils in abscess.
3. Caseous necrosis
Prototype: tuberculosis (granuloma centre); also histoplasmosis.
Mechanism: unique combination of coagulation + partial liquefaction, with lipid-rich mycobacterial wall contributing to a cheese-like texture.
Gross: soft, white, crumbling, cheese-like ("caseous" = Latin caseus, cheese).
Microscopy: amorphous, granular, eosinophilic debris — no ghost cells, surrounded by epithelioid macrophages and Langhans giant cells (the granuloma).
4. Fat necrosis
Prototype: acute pancreatitis (enzymatic); traumatic fat necrosis (breast).
Mechanism: lipases (released from damaged pancreatic acini or trauma) cleave neutral fat in adipocytes into fatty acids + glycerol. Fatty acids bind calcium → insoluble calcium soaps (saponification) → chalky white spots.
Gross: chalky-white opaque deposits in mesenteric/peripancreatic fat; breast lump (trauma).
Microscopy: outlines of adipocytes filled with pale debris, surrounded by inflammatory cells and basophilic calcium deposits.
5. Fibrinoid necrosis
Prototype: immune complex vasculitis (SLE, polyarteritis nodosa), malignant hypertension.
Mechanism: antigen–antibody complexes and fibrin deposit in vessel walls; complement-mediated injury leads to bright pink amorphous material (fibrin + immunoglobulin + necrotic protein).
Microscopy: vessel wall replaced by refractile, brightly eosinophilic, homogeneous smudgy material ("fibrinoid" = fibrin-like).
6. Gangrenous necrosis (covered separately below)
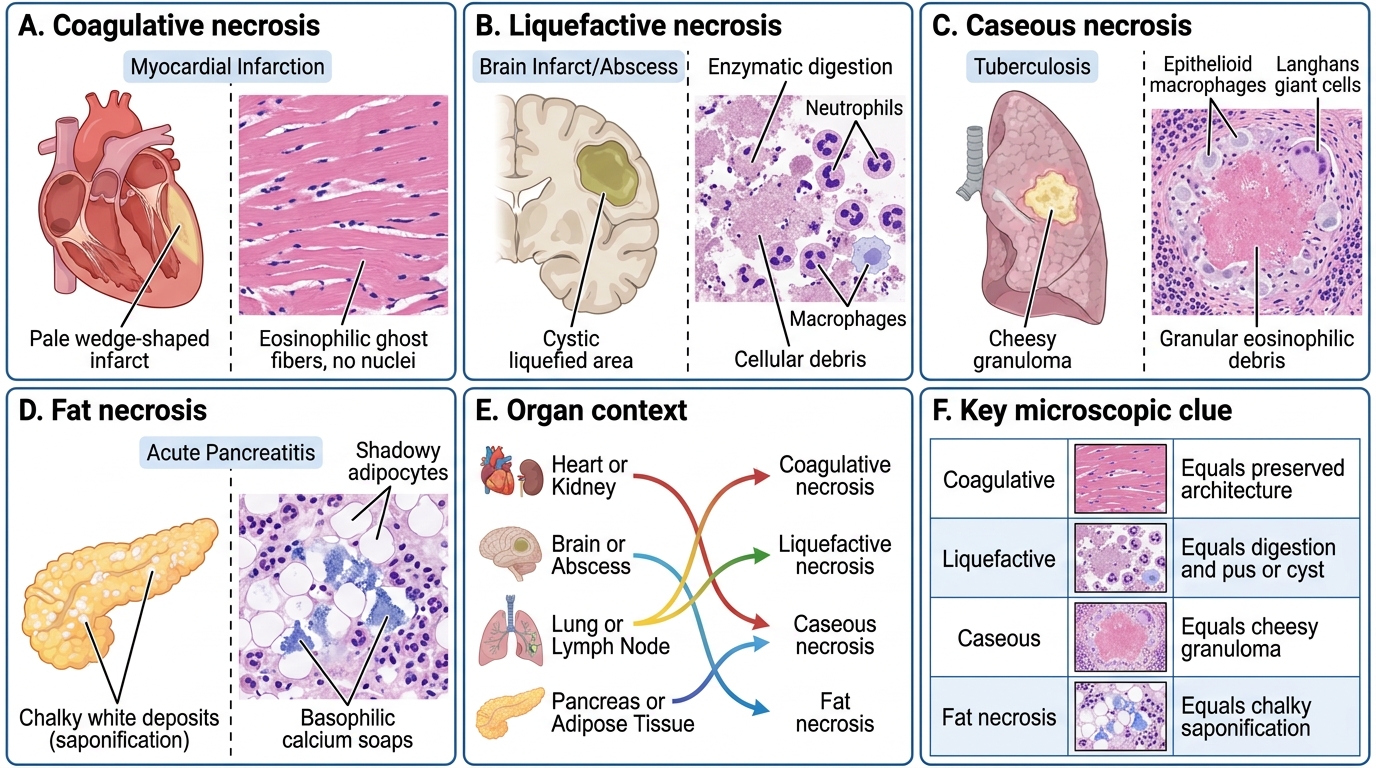
Necrosis Types: Gross, Microscopic, and Organ Context
SELF-CHECK
A 55-year-old man dies 48 hours after an acute anterior myocardial infarction. On microscopy of the infarcted zone, you see eosinophilic cells with preserved outlines but absent nuclei. Which type of necrosis is this?
A. Coagulative necrosis
B. Liquefactive necrosis
C. Caseous necrosis
D. Fibrinoid necrosis
Reveal Answer
Answer: A. Coagulative necrosis
Coagulative necrosis is the hallmark of ischaemic infarction in solid organs (except the brain). The simultaneous denaturation of structural proteins AND lysosomal enzymes preserves the ghost-like cellular architecture for days — hence 'preserved outlines, absent nuclei'. Liquefactive necrosis (option B) occurs in the brain and abscesses where enzymatic digestion dominates. Caseous necrosis (C) shows amorphous debris with no preserved outlines. Fibrinoid necrosis (D) affects vessel walls in immune-mediated disease.
Gangrene — Dry, Wet, and Gas
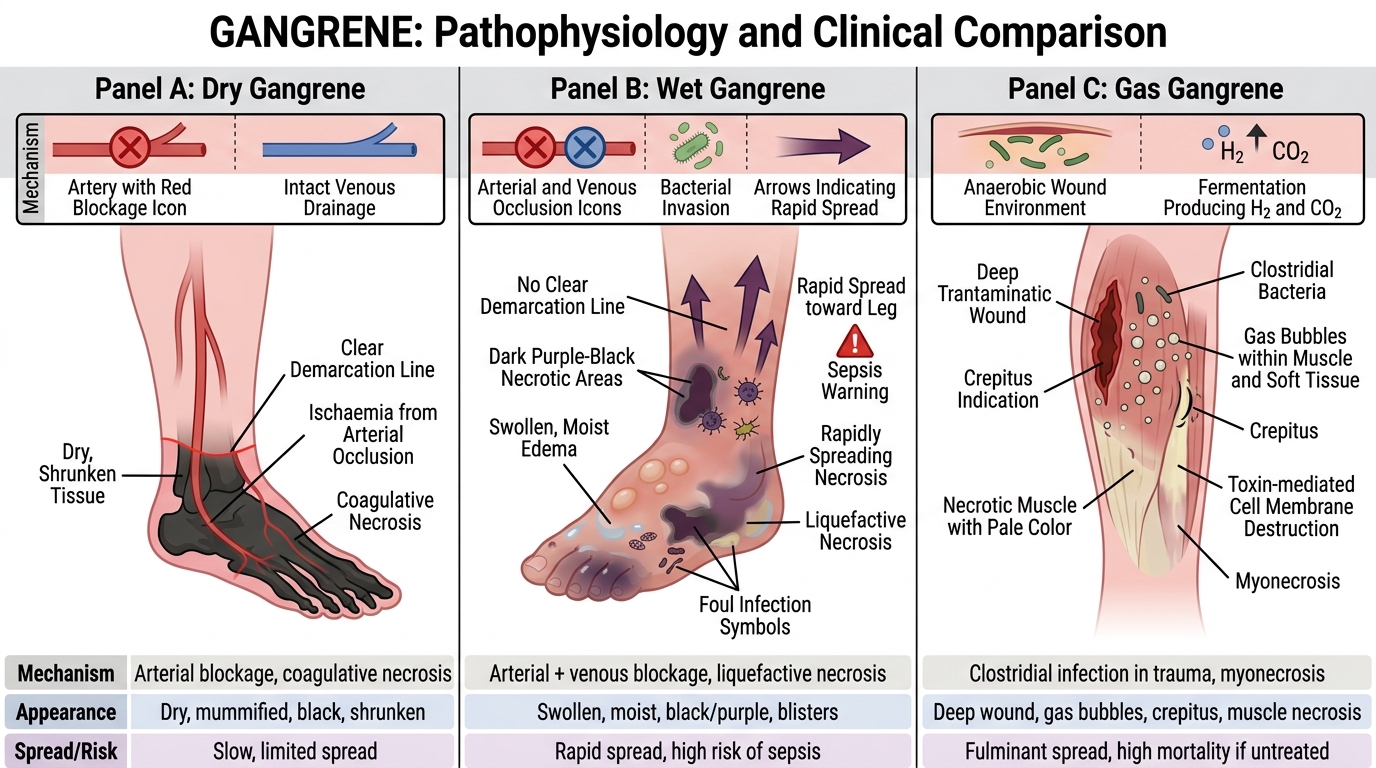
Dry, Wet, and Gas Gangrene
Gangrene is a clinical term for large-scale tissue death, typically of a limb or hollow organ. It is not a separate histological type — it is coagulative or liquefactive necrosis on a macroscopic scale, complicated by putrefactive bacterial infection.
Dry gangrene
• Mechanism: arterial occlusion (atherosclerosis, Buerger's disease) → ischaemia → coagulative necrosis.
• Appearance: limb becomes black, shrunken, dry, mummified (dessication predominates because venous drainage is initially intact).
• Spread: slow; a clear demarcation line (reactive hyperaemia) forms between dead and living tissue.
• No/minimal infection: no pus, no odour initially.
Wet gangrene
• Mechanism: arterial PLUS venous occlusion (or rapid bacterial superinfection of dry gangrene in diabetics) → liquefactive superimposed on coagulative necrosis.
• Appearance: limb is swollen, moist, foul-smelling; skin blisters; no clear line of demarcation.
• Spread: rapid; life-threatening due to systemic sepsis.
• Common context: diabetes (peripheral vascular disease + neuropathy + immunosuppression), internal organs (strangulated bowel, appendicitis with perforation).
Gas gangrene
• Causative organism: Clostridium perfringens (and other clostridia).
• Mechanism: deep contaminated wound (trauma, surgery) → anaerobic clostridial growth → exotoxins (lecithinase/alpha-toxin) destroy cell membranes; fermentation produces H₂ and CO₂ gas in tissues.
• Appearance: crepitus on palpation (gas bubbles under skin); rapidly spreading necrosis; X-ray shows gas in soft tissue.
• Emergency: fasciotomy + antibiotics + hyperbaric O₂; mortality high without prompt debridement.
CLINICAL PEARL
Diabetic foot gangrene is almost always wet because autonomic neuropathy causes arteriovenous shunting (impaired vasoconstriction) and peripheral oedema — venous stasis is added to arterial insufficiency. Add immune dysfunction and peripheral neuropathy (unnoticed trauma), and you have the perfect storm. A dry-appearing diabetic foot can convert to wet gangrene within 24–48 hours if infected — always examine for warmth, fluctuance, and crepitus even when the foot looks 'dry and black'.