Page 6 of 25
PA3.3 | Chronic & Granulomatous Inflammation — SDL Guide
Learning Objectives
- Define chronic inflammation and distinguish it from acute inflammation by duration, cellular composition, and tissue consequences.
- List the principal causes of chronic inflammation including persistent infection, autoimmunity, foreign body, and unresolved acute episodes.
- Identify and describe the key cells of chronic inflammation — macrophages (M1/M2 activation, epithelioid transformation), lymphocytes, plasma cells, eosinophils, and mast cells — and the macrophage–lymphocyte cross-talk that sustains the response.
- Contrast non-specific (diffuse) chronic inflammation with granulomatous inflammation.
- Define a granuloma, describe its cellular components (epithelioid histiocytes, Langhans and foreign-body giant cells), and explain Th1/IFN-γ/TNF-driven formation.
- Distinguish caseating from non-caseating granulomas and relate each pattern to specific disease causes.
- Enumerate the classic causes of granulomatous inflammation and identify distinguishing histological or clinical features.
- Describe the systemic effects of inflammation: fever (pyrogens), acute-phase proteins (CRP, fibrinogen), and leucocytosis.
INSTRUCTIONS
Chronic inflammation is the pathological substrate of tuberculosis, sarcoidosis, Crohn disease, and dozens of other conditions you will encounter throughout your clinical years. Understanding why a granuloma forms — and what its cellular architecture tells you about the causative agent — is the single most clinically translatable skill in general pathology. This module builds directly on Cluster G1 (Acute Inflammation) and feeds into your understanding of infections, autoimmunity, and systemic disease.
References
- Robbins & Kumar: Basic Pathology, 11th ed., Ch 2 (Inflammation and Repair) (textbook)
- Harsh Mohan: Textbook of Pathology, 8th ed., Ch 5 (Chronic Inflammation) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 35-year-old man presents with a 3-month history of low-grade fever, night sweats, and a 5 kg weight loss. Chest X-ray shows a right upper lobe consolidation with a central lucency. His sputum smear is AFB-positive. At autopsy of untreated disease, the pathologist finds a lung nodule with a chalky-white centre that crumbles like dry cheese.
That crumbling centre — caseous necrosis inside a granuloma — is the microscopic signature of tuberculosis. By the end of this module you will know exactly why the immune system builds that structure, what cells compose it, and how to distinguish it from the granulomas of sarcoidosis, Crohn disease, or a splinter in the finger.
WHY THIS MATTERS
Granulomatous inflammation underpins a dozen high-yield diseases you will diagnose and manage as a physician:
- Caseating granulomas → TB (India bears the world's highest burden), leprosy, deep fungal infections
- Non-caseating granulomas → sarcoidosis, Crohn disease, foreign-body reactions, cat-scratch disease
- Misidentifying the granuloma type or missing the causative organism delays treatment, sometimes fatally.
Chronic inflammation also explains the systemic features — fever, weight loss, elevated ESR/CRP — that you will use to monitor disease activity and treatment response.
RECALL
Before proceeding, confirm your foundation from Cluster G1:
- What is the cardinal cellular hallmark of acute inflammation?
- Name the five classical signs of acute inflammation and their vascular basis.
- What is the difference between exudate and transudate?
- What happens when the injurious agent is not eliminated at the end of acute inflammation?
If question 4 is unclear, re-read the resolution section of G1-SDL1 before continuing — chronic inflammation begins exactly where acute inflammation fails to resolve.
Defining Chronic Inflammation
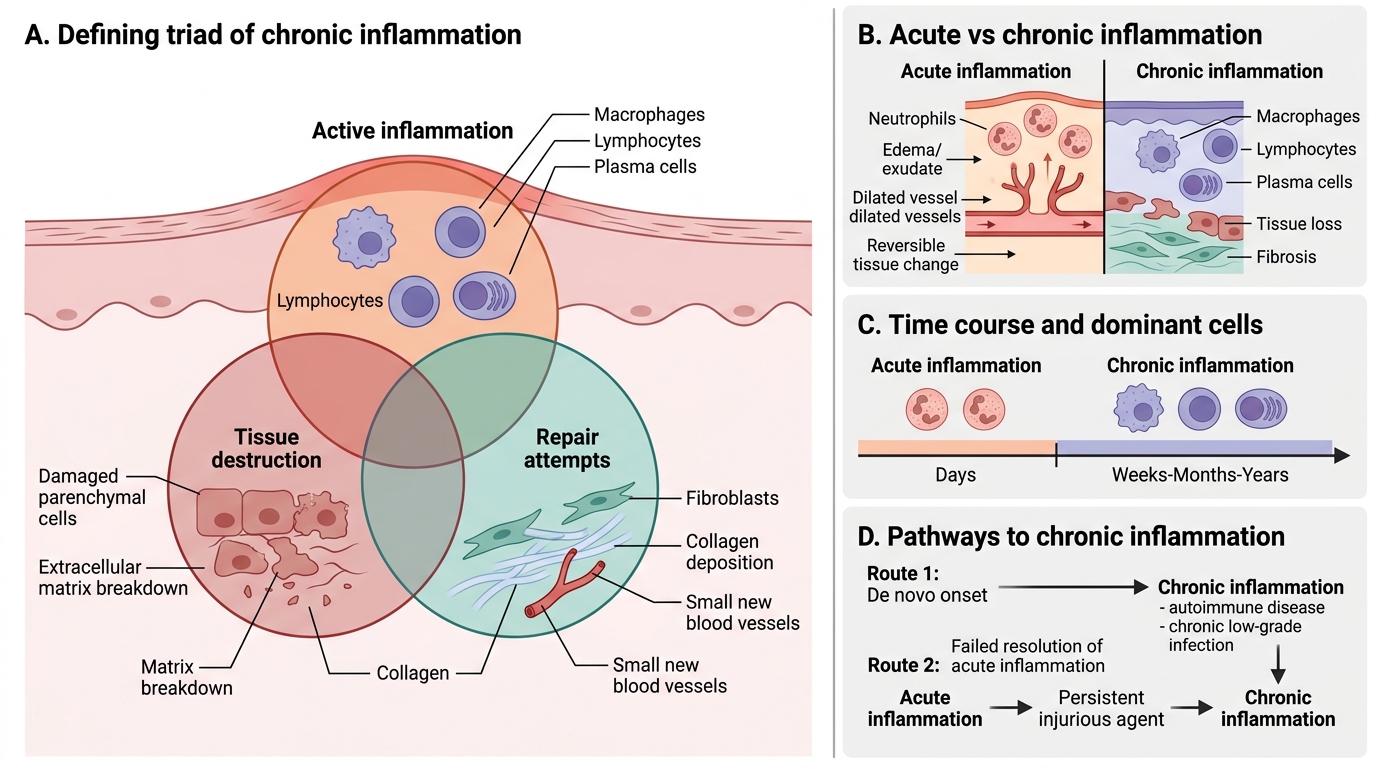
Chronic Inflammation: Defining Triad and Origins
Chronic inflammation is a prolonged inflammatory response (weeks to months) in which active inflammation, tissue destruction, and repair attempts occur simultaneously. This is the defining triad — not merely long duration.
Key contrasts with acute inflammation:
| Feature | Acute | Chronic |
|---|---|---|
| Duration | Days | Weeks–months–years |
| Dominant cells | Neutrophils | Macrophages, lymphocytes, plasma cells |
| Tissue outcome | Exudation; often reversible | Destruction + fibrosis; often permanent |
| Vascular changes | Prominent (rubor, calor) | Subtle |
Chronic inflammation may arise:
1. De novo — directly, without a preceding acute episode (e.g., autoimmune diseases, chronic low-grade infections)
2. After failed resolution of acute inflammation — when the injurious agent persists, repeated or prolonged acute episodes transition to chronic
> In clinical practice, chronic inflammation is the tissue-level explanation for why a patient with untreated TB or Crohn disease continues to deteriorate months after the initial event.
Causes of Chronic Inflammation
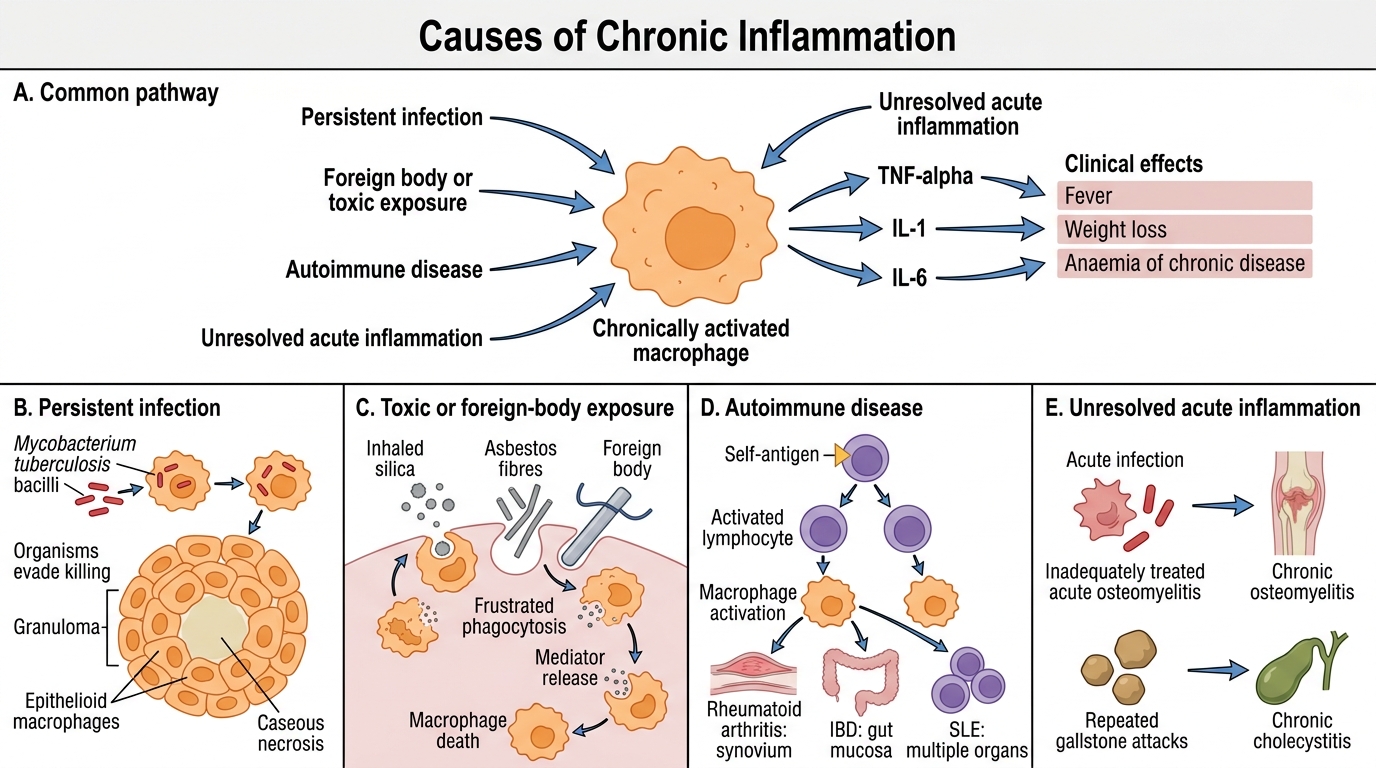
Causes of Chronic Inflammation
Four major mechanisms sustain chronic inflammation:
- Persistent infections — organisms that evade phagocytic killing (Mycobacterium tuberculosis, M. leprae, Treponema pallidum, Helicobacter pylori, certain fungi). The immune system cannot eradicate them; instead it walls them off, generating granulomas.
- Prolonged toxic or foreign-body exposure — silica dust (silicosis), asbestos fibres (asbestosis), suture material, implants. Macrophages cannot digest these agents and die, releasing mediators that recruit more macrophages in a futile cycle.
- Autoimmune diseases — self-antigens continuously activate lymphocytes and macrophages (rheumatoid arthritis — synovium; SLE — multiple organs; inflammatory bowel disease — gut mucosa).
- Unresolved acute inflammation — e.g., chronic osteomyelitis following inadequately treated acute osteomyelitis; chronic cholecystitis following repeated gallstone-induced attacks.
Clinical link: The systemic feature common to all these settings — persistent fever, weight loss, anaemia of chronic disease — reflects sustained cytokine release (TNF-α, IL-1, IL-6) from chronically activated macrophages.
Cells of Chronic Inflammation
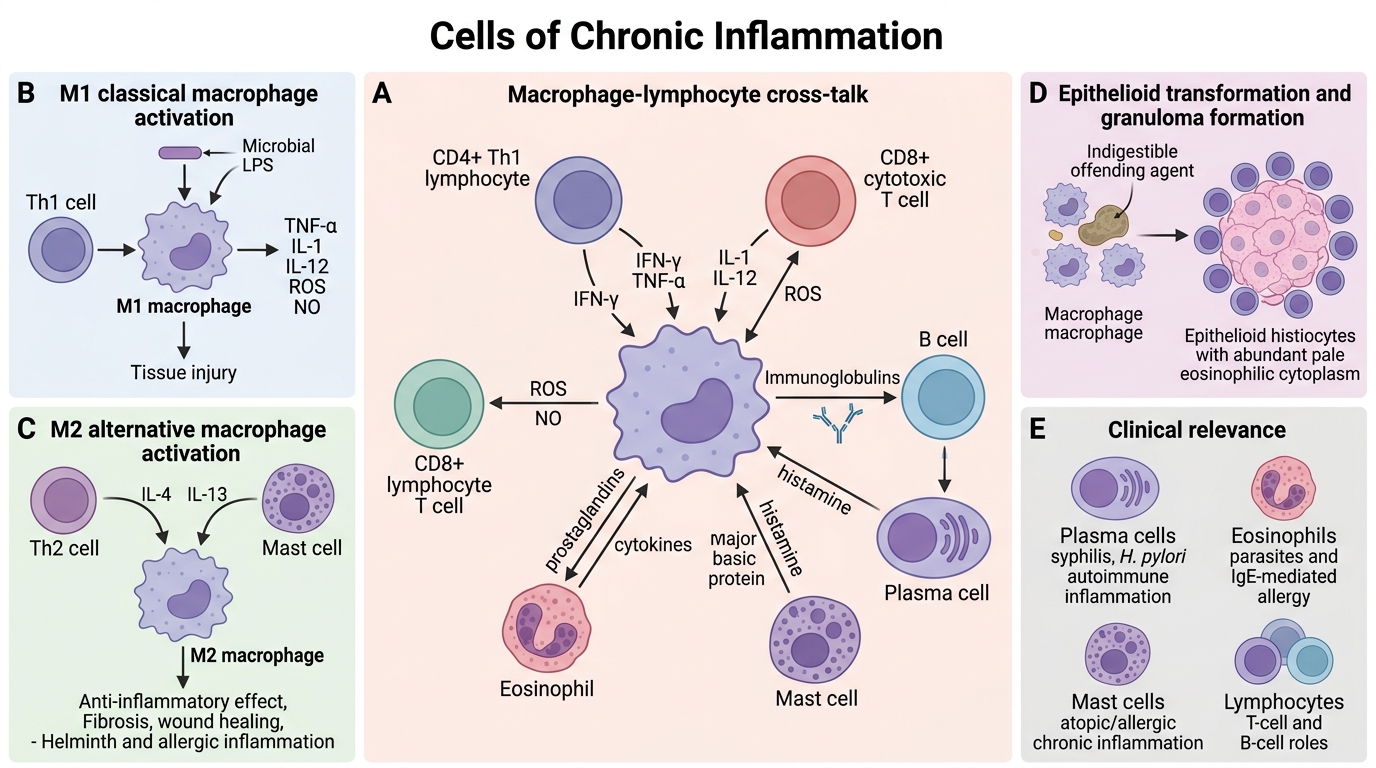
Macrophages are the dominant orchestrators of chronic inflammation.
- Activated macrophages (M1 — classical activation): Triggered by IFN-γ (from Th1 cells) and microbial products (LPS). Produce TNF-α, IL-1, IL-12, reactive oxygen species (ROS), nitric oxide. Microbicidal, pro-inflammatory. Responsible for tissue damage in chronic lesions.
- Alternatively activated macrophages (M2): Triggered by IL-4, IL-13 (from Th2 cells and mast cells). Anti-inflammatory; promote fibrosis and wound healing. Elevated in helminth infections and allergic inflammation.
- Epithelioid transformation: When macrophages cannot digest the offending agent, they develop abundant pale eosinophilic cytoplasm and vesicular nuclei — resembling epithelial cells. These are epithelioid histiocytes (the defining cell of a granuloma).
Other cells:
- Lymphocytes (T and B): CD4+ Th1 cells activate macrophages via IFN-γ; CD8+ cytotoxic cells kill infected cells; B cells mature into plasma cells locally.
- Plasma cells: Terminally differentiated B cells; produce immunoglobulins. Seen prominently in syphilis, H. pylori gastritis, and autoimmune inflammation.
- Eosinophils: Dominant in parasitic infections and IgE-mediated (allergic) inflammation. Contain major basic protein, toxic to parasites and host tissue alike.
- Mast cells: Release histamine, prostaglandins, and cytokines. Bridge innate and adaptive responses; prominent in atopic and allergic chronic inflammation.
Macrophage–lymphocyte cross-talk: CD4+ Th1 cells secrete IFN-γ → activates macrophages → macrophages secrete IL-12 → further Th1 polarisation. This self-amplifying loop drives and perpetuates granuloma formation.
Cells of Chronic Inflammation
SELF-CHECK
Which cytokine produced by Th1 lymphocytes is MOST responsible for classical (M1) macrophage activation in chronic inflammation?
A. Interleukin-4 (IL-4)
B. Interleukin-13 (IL-13)
C. Interferon-gamma (IFN-γ)
D. Interleukin-10 (IL-10)
Reveal Answer
Answer: C. Interferon-gamma (IFN-γ)
IFN-γ secreted by Th1 CD4+ cells is the principal signal that drives classical (M1) macrophage activation. IL-4 and IL-13 drive alternative (M2) activation and are characteristic of Th2 responses and helminth immunity. IL-10 is an anti-inflammatory cytokine.