Page 6 of 24
PA6.2-3 | Molecular Basis of Cancer & Carcinogenesis — SDL Guide
Learning Objectives
- Explain the hallmarks of cancer and the molecular events underlying each hallmark
- Distinguish oncogenes from tumour suppressor genes using prototypical examples (RAS, MYC, RB, TP53)
- Apply Knudson's two-hit hypothesis to hereditary versus sporadic cancers
- Describe the roles of BCL2, DNA repair genes (MMR, BRCA), and epigenetic/microRNA dysregulation in carcinogenesis
- Classify carcinogens (chemical, radiation, microbial/viral) and outline the mechanism of each class
- Trace the multistep clonal evolution model from a single initiated cell to invasive carcinoma
INSTRUCTIONS
Cancer is not one disease but a family of diseases united by a common principle: cells escape the controls that govern normal growth. Molecular pathology has decoded the grammar of this escape — and that grammar now drives every targeted therapy, screening test, and cancer-risk calculation you will encounter in clinical practice. This module builds that grammar systematically, moving from the hallmarks of cancer through cancer genes, carcinogens, and the multistep model, giving you a framework that will serve you from the ward to the exam hall.
References
- Robbins & Kumar: Basic Pathology, 11th ed., Ch 5 — Neoplasia (textbook)
- Harsh Mohan: Textbook of Pathology, 8th ed., Ch 8 — Neoplasia (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 28-year-old non-smoker with no family history is diagnosed with lung adenocarcinoma harbouring an EGFR exon-19 deletion. Her oncologist prescribes erlotinib — a pill, not chemotherapy — and her tumour shrinks by 70% within six weeks. Why does a mutation in a growth-factor receptor make a tumour exquisitely sensitive to one targeted drug while making it almost invisible to conventional chemotherapy? The answer lies in the molecular architecture of cancer itself.
WHY THIS MATTERS
For a clinician, understanding cancer biology is not an academic luxury. Every histopathology report you read references molecular markers. Every consent discussion about hereditary cancer risk invokes genes like BRCA1 and TP53. Targeted therapies — now the backbone of oncology — are only intelligible if you understand the molecular targets they were designed to hit. The NMC CBME framework explicitly links PA6.2–PA6.3 to the competency of applying molecular pathology in clinical reasoning.
RECALL
Before we proceed, activate what you already know:
- From Year-1 Biochemistry: What is a proto-oncogene? How does a point mutation differ from a gene amplification?
- From Year-1 Physiology: What is the cell cycle, and at what checkpoints does the cell assess DNA integrity?
- From Year-1 Anatomy/Histology: What is the basement membrane, and what does 'invasion through the basement membrane' mean structurally?
If any of these feel unclear, spend three minutes with a quick review — they are the scaffolding on which this module hangs.
The Hallmarks of Cancer
Cancer is a disease of dysregulated cell behaviour. Hanahan & Weinberg's framework of hallmarks of cancer organises this dysregulation into ten interrelated capabilities that a cancer cell must acquire:
- Self-sufficiency in growth signals — cancer cells generate their own mitogenic stimuli (autocrine loops, activating mutations in receptors such as EGFR).
- Insensitivity to growth-inhibitory signals — normal brakes (RB, TGF-β pathway) are disabled.
- Evading apoptosis — cells block programmed death (BCL2 overexpression, TP53 loss).
- Limitless replicative potential — telomerase is reactivated, removing the Hayflick limit.
- Sustained angiogenesis — tumours >1–2 mm recruit new vessels via VEGF.
- Tissue invasion and metastasis — loss of E-cadherin, matrix metalloproteinase upregulation.
- Reprogrammed energy metabolism (Warburg effect) — aerobic glycolysis even in the presence of oxygen; supports rapid biosynthesis.
- Evading immune destruction — PD-L1 upregulation, loss of MHC I, immunosuppressive microenvironment.
- Genome instability and mutation — mutations in DNA repair machinery accelerate evolution.
- Tumour-promoting inflammation — NF-κB-driven cytokines supply growth factors and pro-survival signals.
No single mutation confers all ten hallmarks. They are acquired progressively — the essence of the multistep model covered below.
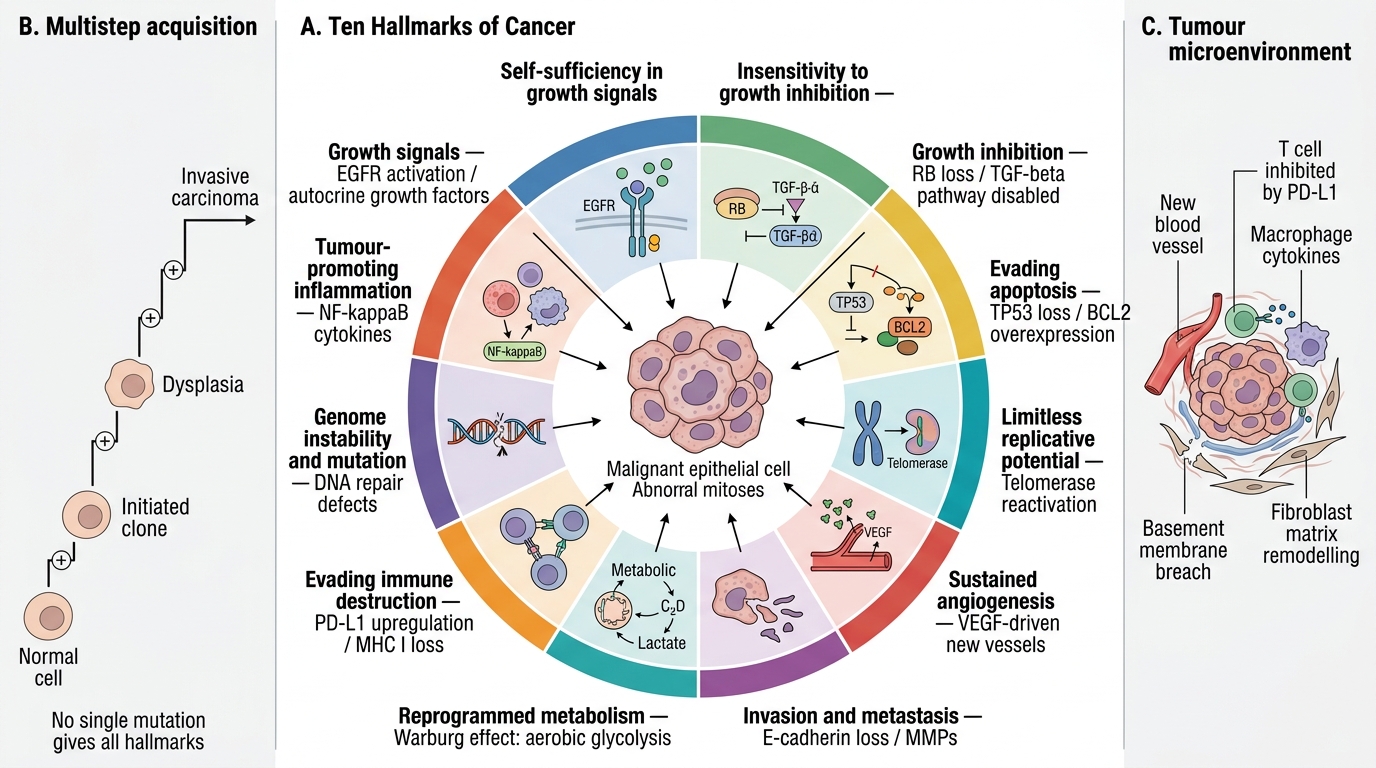
The Ten Hallmarks of Cancer
SELF-CHECK
A 55-year-old man's colon cancer cells are shown to proliferate even in serum-free medium and are resistant to TGF-β-induced growth arrest. Which two hallmarks of cancer are directly illustrated?
A. Limitless replication and sustained angiogenesis
B. Self-sufficiency in growth signals and insensitivity to growth-inhibitory signals
C. Evading apoptosis and genome instability
D. Warburg effect and immune evasion
Reveal Answer
Answer: B. Self-sufficiency in growth signals and insensitivity to growth-inhibitory signals
Proliferation in serum-free medium = self-sufficiency in growth signals (no external mitogens needed). Resistance to TGF-β arrest = insensitivity to growth-inhibitory signals. TGF-β normally halts progression through the G1/S checkpoint via upregulation of CDK inhibitors.
Oncogenes and Proto-oncogenes
A proto-oncogene is a normal cellular gene that promotes growth, differentiation, or survival. When mutated or amplified to become constitutively active, it becomes an oncogene — functioning like a stuck accelerator.
Mechanisms of activation:
• Point mutation — single amino-acid change locks a protein in the active state (classic: RAS Gly12→Val)
• Gene amplification — extra copies of the gene → excess protein (HER2/ERBB2 in breast cancer; MYC in neuroblastoma)
• Chromosomal translocation — brings a gene under a strong promoter or creates a novel fusion protein (BCR-ABL in CML; MYC under Ig promoter in Burkitt lymphoma)
• Insertional mutagenesis — retroviral DNA integrates upstream of a proto-oncogene
Key examples to memorise:
| Gene | Normal function | Cancer type | Mechanism |
|---|---|---|---|
| RAS (KRAS, NRAS, HRAS) | GTPase signal transducer | Pancreas, colon, lung | Point mutation (Gly12); GTPase activity lost, always-ON |
| MYC | Transcription factor, drives cell-cycle entry | Burkitt lymphoma, neuroblastoma | Translocation or amplification |
| HER2/ERBB2 | Receptor tyrosine kinase | Breast, gastric | Amplification (trastuzumab target) |
| ABL | Non-receptor tyrosine kinase | CML | t(9;22) BCR-ABL fusion — imatinib target |
Important principle: oncogene mutations are dominant — one mutant allele is sufficient because the protein is gain-of-function.
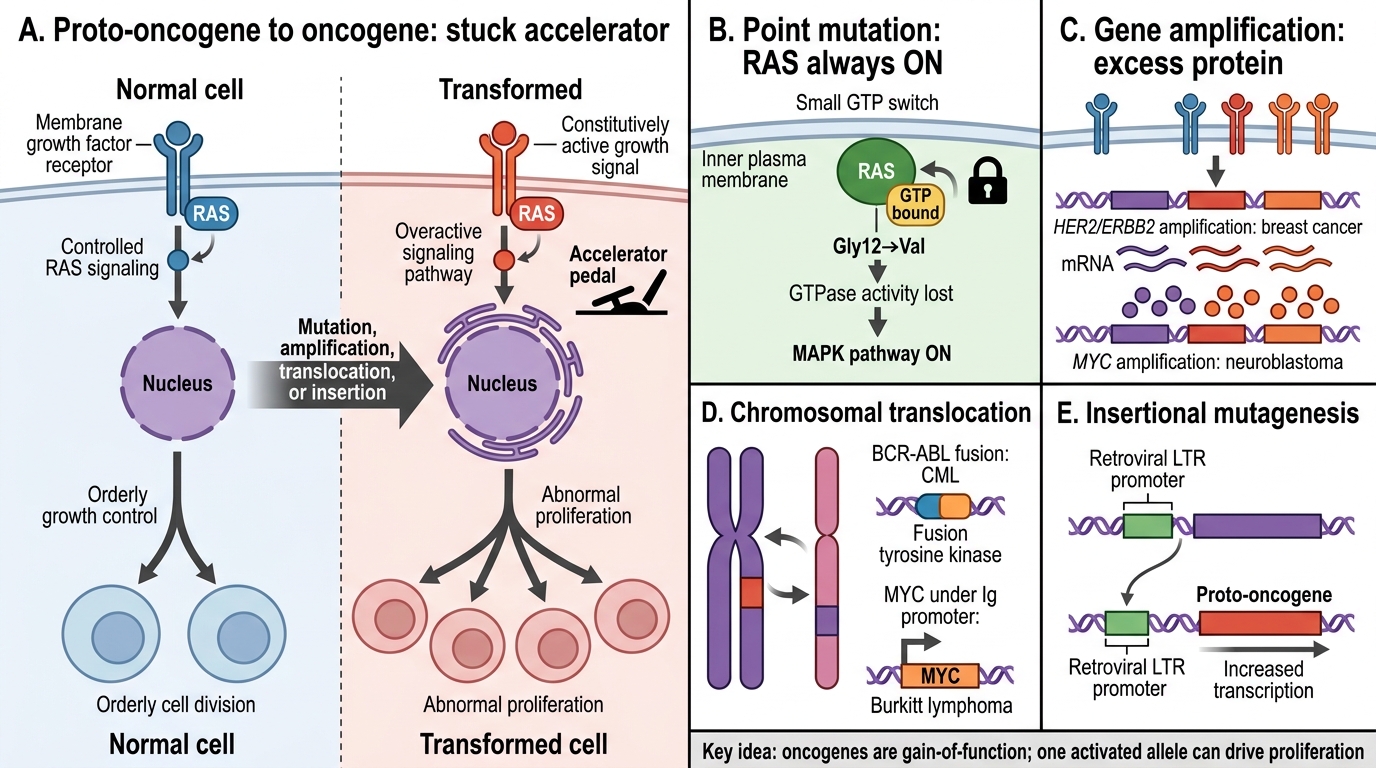
Activation of Proto-oncogenes into Oncogenes
Tumour Suppressor Genes
Tumour suppressor genes (TSGs) encode proteins that restrain proliferation, promote apoptosis, or maintain genomic fidelity. Unlike oncogenes, TSGs must lose both alleles — they are recessive at the cellular level (though dominant at the family/pedigree level in hereditary syndromes).
Knudson's two-hit hypothesis: Knudson (1971) compared retinoblastoma rates in hereditary vs sporadic cases. He proposed:
• Hereditary: first hit inherited in the germline (present in every cell), second hit is a somatic mutation → early-onset, bilateral/multifocal tumours
• Sporadic: both hits must occur somatically in the same cell → later onset, unilateral
This model now applies to RB, TP53, APC, BRCA1/2, and many others.
Key TSGs:
| Gene | Protein/Function | Cancer associations |
|---|---|---|
| RB (retinoblastoma) | pRB: binds E2F, blocks S-phase entry when hypophosphorylated | Retinoblastoma, osteosarcoma; functionally inactivated in most human cancers |
| TP53 | p53: transcription factor — halts cell cycle, induces DNA repair or apoptosis after genotoxic stress; 'guardian of the genome' | >50% of all human cancers; Li-Fraumeni syndrome |
| APC | Destroys β-catenin; suppresses Wnt pathway | Familial adenomatous polyposis (FAP), sporadic colon cancer |
| BRCA1/2 | Homologous recombination DNA repair | Hereditary breast/ovarian cancer |
Key distinction: TSG inactivation is loss-of-function; the gene product is absent or non-functional.
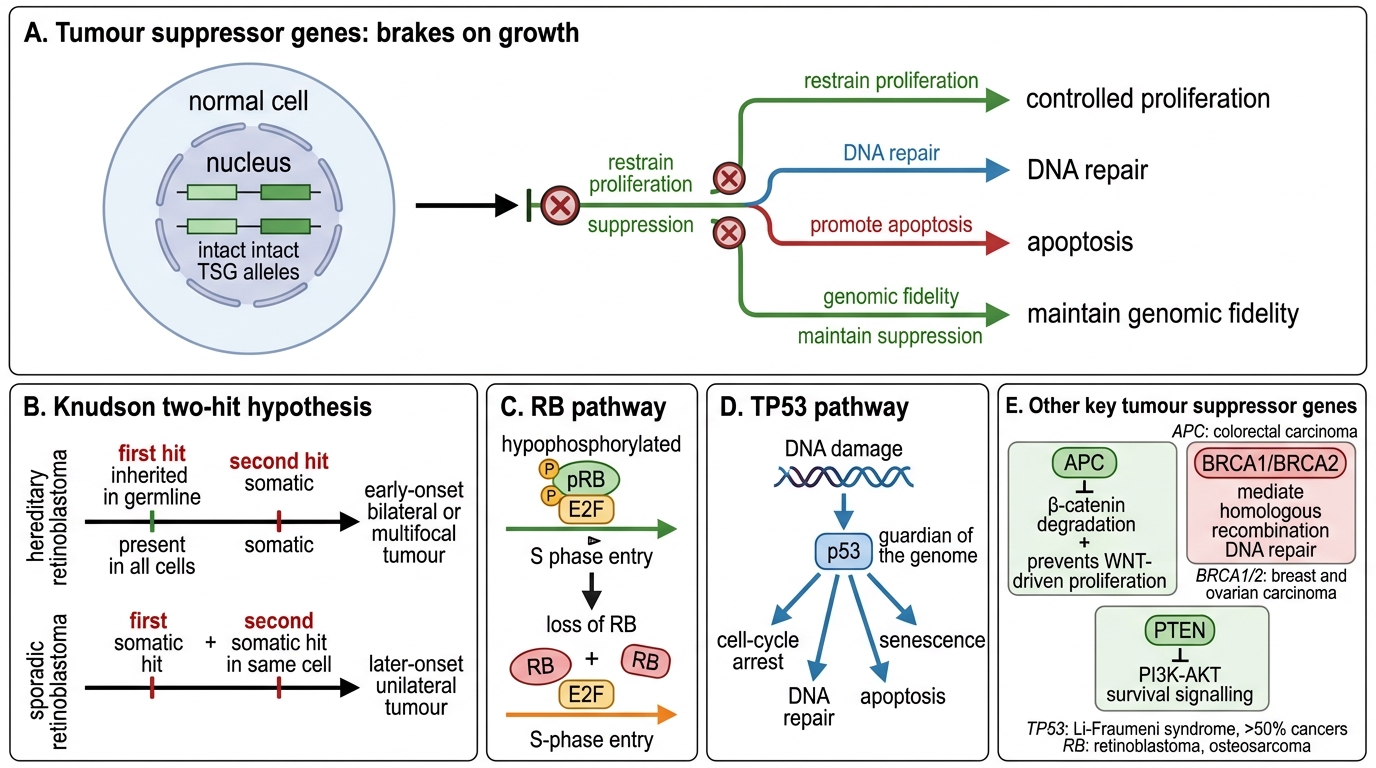
Tumour Suppressor Genes and the Two-Hit Model
SELF-CHECK
A child develops bilateral retinoblastomas at age 18 months. Molecular analysis shows a germline RB mutation on one allele and a somatic deletion of the second allele in the tumour. Which statement BEST explains this finding?
A. The germline mutation is the second hit; the somatic deletion is the first hit
B. Both hits are somatic; germline mutations do not predispose to retinoblastoma
C. The germline mutation is the first hit; loss of the remaining wild-type allele is the second hit, confirming Knudson's model
D. One hit is sufficient when the mutation is inherited; no second hit is required
Reveal Answer
Answer: C. The germline mutation is the first hit; loss of the remaining wild-type allele is the second hit, confirming Knudson's model
Knudson's two-hit hypothesis states that both copies of a tumour suppressor gene must be inactivated. In hereditary cases, the first hit is inherited (present in all cells), so only one somatic event (the second hit) is needed — explaining early onset and bilaterality. The germline mutation is always the first hit in this context.