Page 7 of 24
PA6.2-3 | Molecular Basis of Cancer & Carcinogenesis — SDL Guide (Part 2)
Apoptosis Regulators and DNA Repair Genes
Apoptosis Regulators and DNA Repair Genes in Neoplasia
Apoptosis regulators:
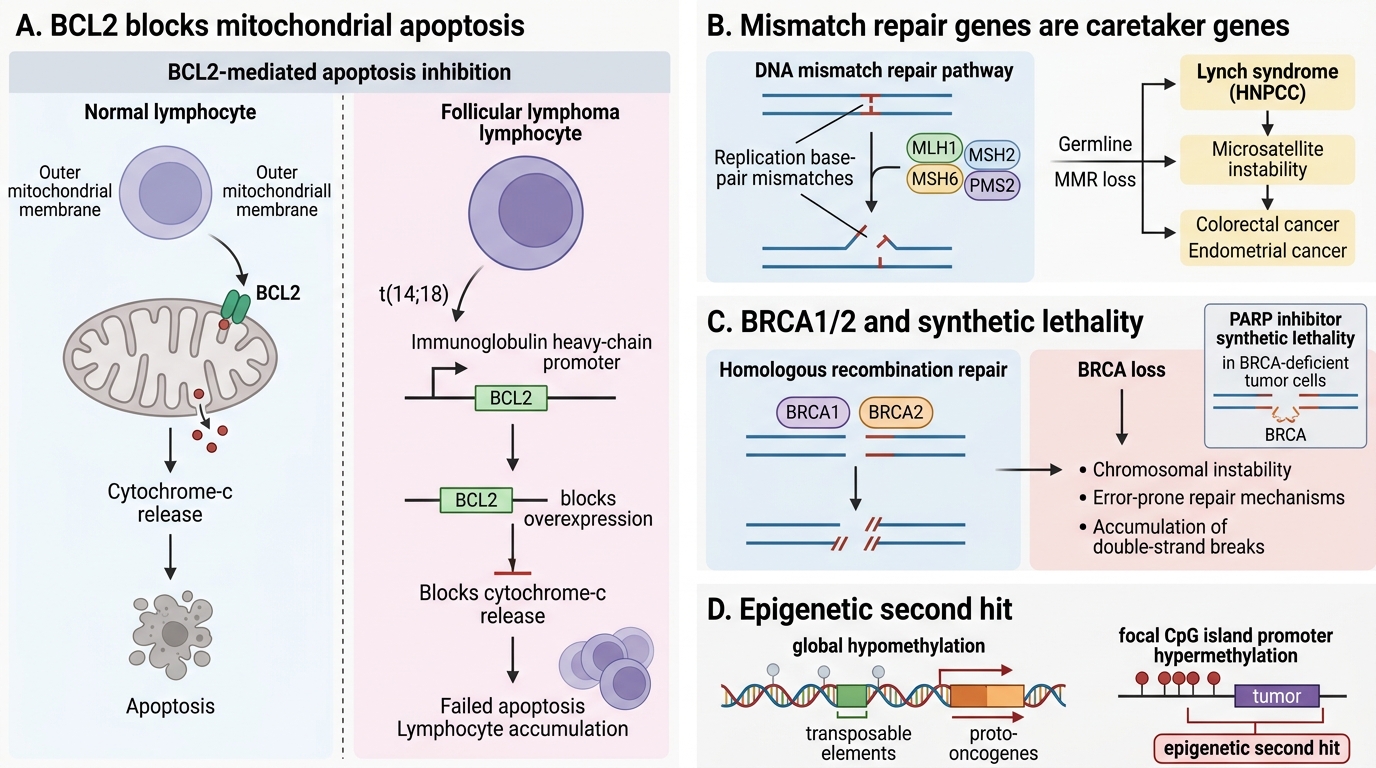
BCL2 (B-cell lymphoma 2) is the prototype anti-apoptotic protein. It resides on the outer mitochondrial membrane and prevents cytochrome-c release. In follicular lymphoma, the t(14;18) translocation places BCL2 under the immunoglobulin heavy-chain promoter → constitutive overexpression → lymphocytes cannot die → accumulate as low-grade lymphoma. BCL2 does NOT drive proliferation; it prevents death — an important distinction from oncogenes.
DNA repair genes — 'caretaker' genes:
When DNA repair fails, mutation rate rises and clonal evolution accelerates.
- Mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2) — correct base-pair mismatches after replication. Germline loss → Lynch syndrome (hereditary non-polyposis colorectal cancer, HNPCC): early-onset colorectal and endometrial cancers with microsatellite instability (MSI) as the molecular signature. Somatic MMR loss also occurs in sporadic colon, gastric, and endometrial cancers.
- BRCA1/2 — as noted above, enable homologous recombination. Loss → error-prone repair, chromosomal instability, double-strand break accumulation. BRCA-deficient tumours are exquisitely sensitive to PARP inhibitors (synthetic lethality).
Epigenetic alterations:
Cancer genomes are globally hypomethylated (activating transposable elements and proto-oncogenes) yet show focal hypermethylation of TSG promoter CpG islands, silencing them without mutation — the so-called 'epigenetic second hit'.
MicroRNAs (miRNAs):
miRNAs are small non-coding RNAs that post-transcriptionally repress target mRNAs. OncomiRs (e.g., miR-21) suppress TSGs; tumour-suppressor miRNAs (e.g., miR-15/16) suppress BCL2. miRNA dysregulation is now a recognised mechanism of carcinogenesis and a potential biomarker.
CLINICAL PEARL
Practical yield of MMR/MSI testing: Universal MMR immunohistochemistry (or MSI-PCR) is now standard on all newly diagnosed colorectal cancers. MSI-high status predicts: (1) Lynch syndrome workup needed if patient is <70 years, (2) poor response to standard 5-FU adjuvant chemotherapy, but (3) excellent response to immune checkpoint inhibitors (pembrolizumab). Understanding the biology directly changes management.
Multistep Carcinogenesis and Clonal Evolution
No single mutation transforms a normal cell into a fully malignant cancer. Multistep carcinogenesis is the sequential acquisition of mutations over years to decades, each conferring a selective growth advantage — Darwinian evolution within a tissue.
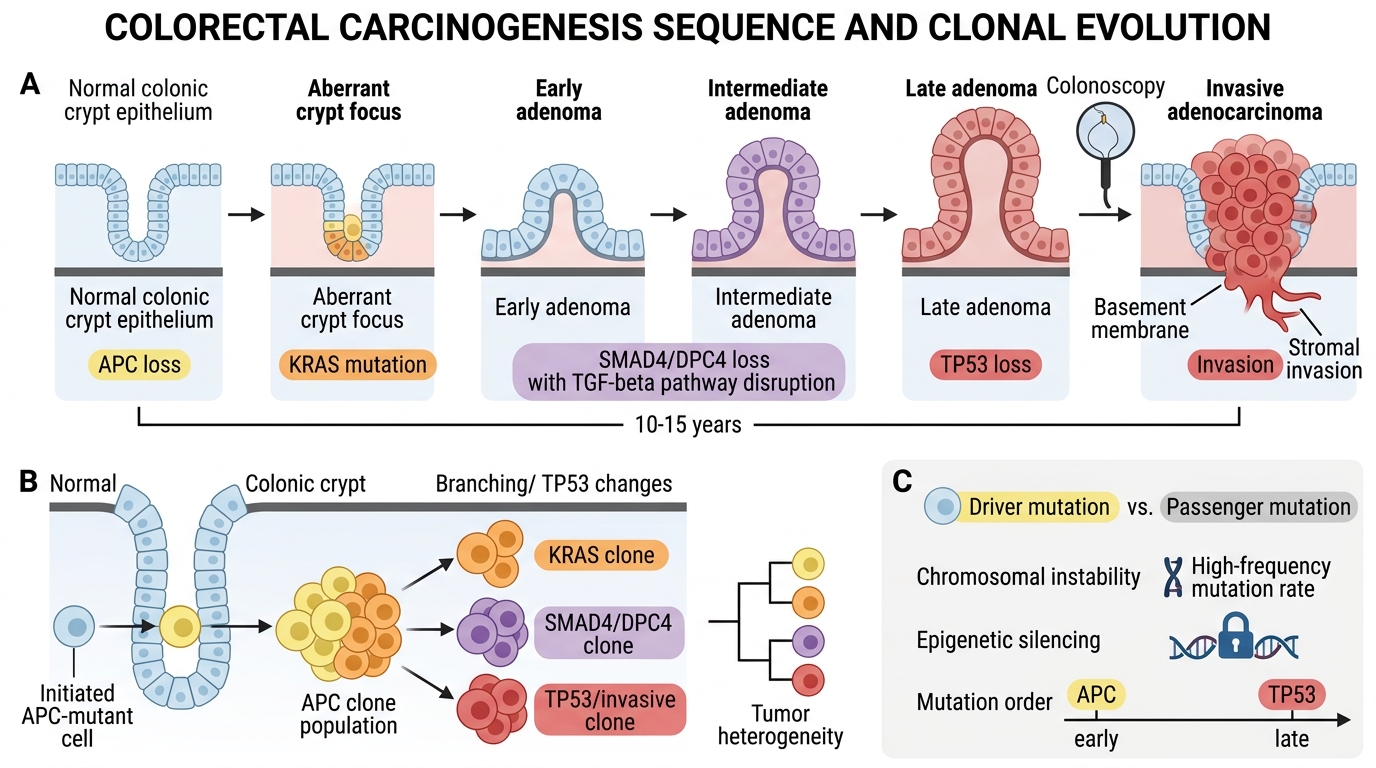
The colon provides the best-characterised model (Fearon–Vogelstein sequence):
APC loss → aberrant crypt foci → early adenoma (KRAS mutation) → intermediate adenoma (SMAD4/DPC4 loss, TGF-β pathway) → late adenoma (TP53 loss) → invasive adenocarcinoma
This sequence typically takes 10–15 years, providing a window for screening colonoscopy to interrupt progression.
Key principles:
• The order of mutations matters — APC loss is the obligate early event; TP53 loss is typically late
• 'Driver mutations' provide selective advantage; 'passenger mutations' accumulate but are not necessary
• Clonal evolution: a single initiated cell with a growth advantage outcompetes neighbours, then subclones with further mutations arise within that clone — the basis of tumour heterogeneity
• Epigenetic changes and chromosomal instability (CIN) amplify the mutation rate at each step
Multistep Carcinogenesis and Clonal Evolution in Colorectal Cancer
SELF-CHECK
A 45-year-old man undergoes surveillance colonoscopy for familial adenomatous polyposis (FAP). A 1.2 cm tubular adenoma is removed. Sequencing reveals: APC germline mutation (inherited) + somatic KRAS Gly12Val mutation. According to the multistep model, what is the MOST likely next molecular event if this adenoma were to progress toward carcinoma?
A. Somatic APC second-hit mutation
B. KRAS amplification
C. Loss of SMAD4 or TP53 function
D. BCL2 translocation
Reveal Answer
Answer: C. Loss of SMAD4 or TP53 function
In the Fearon–Vogelstein sequence, APC loss and KRAS mutation represent early events. The intermediate-to-late adenoma transitions are driven by SMAD4 loss (disabling TGF-β growth arrest) and ultimately TP53 loss (abrogating the DNA-damage checkpoint), enabling invasive behaviour. BCL2 translocation is associated with follicular lymphoma, not colorectal carcinogenesis.
Chemical Carcinogenesis
Chemical Carcinogenesis: Initiation, Promotion, and DNA Injury
Chemical carcinogenesis proceeds in two separable stages:
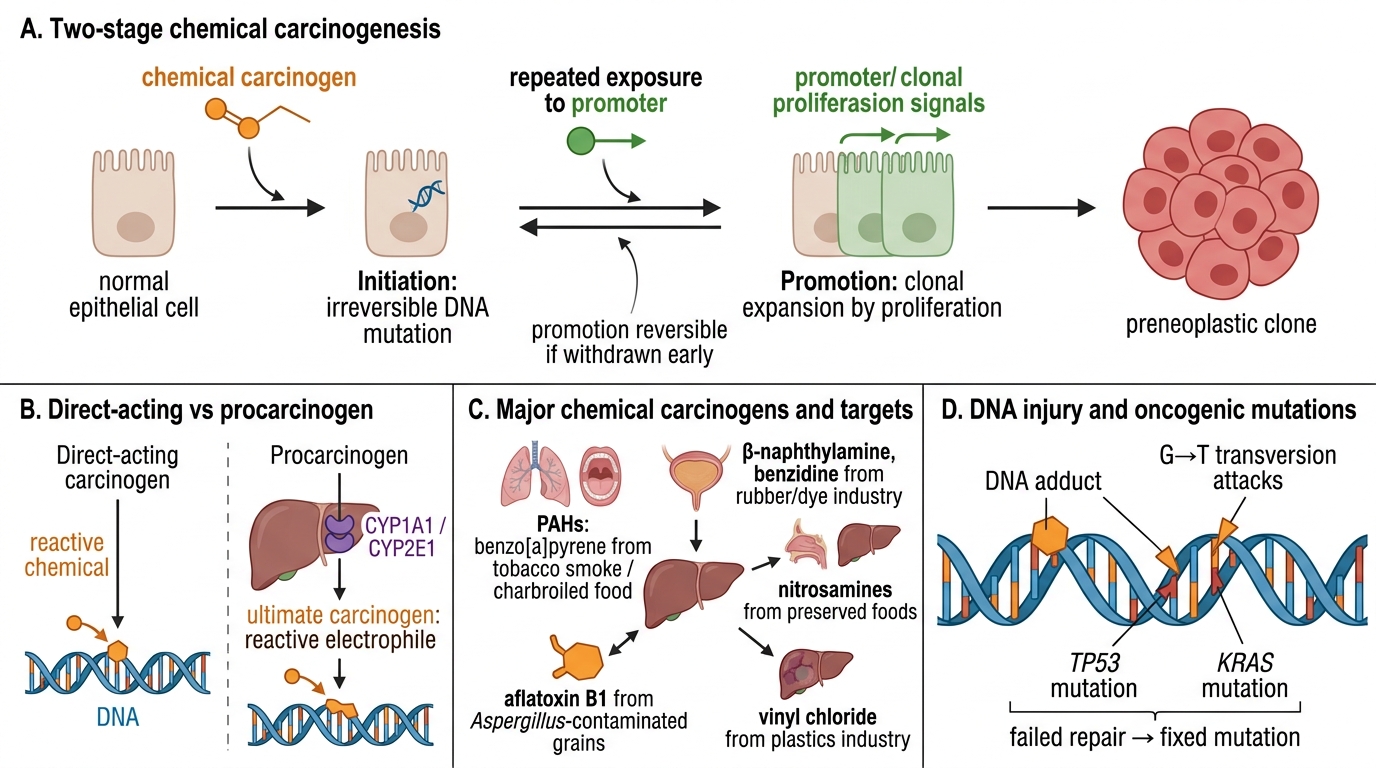
Initiation: an irreversible, heritable DNA mutation induced by a chemical carcinogen. The cell is 'initiated' but not yet transformed.
Promotion: repeated, prolonged exposure to a promoter (not itself mutagenic) causes clonal expansion of the initiated cell by stimulating proliferation. Promotion is reversible if the promoter is withdrawn early.
Direct-acting vs procarcinogens:
• Direct-acting carcinogens (e.g., alkylating agents, nitrogen mustard, cyclophosphamide) react with DNA without metabolic activation.
• Procarcinogens require metabolic activation by cytochrome P450 enzymes (CYP1A1, CYP2E1) in the liver or target organ to form the ultimate carcinogen — the reactive electrophile that attacks DNA.
Key examples:
| Carcinogen | Source | Target organ | Mechanism/Note |
|---|---|---|---|
| Polycyclic aromatic hydrocarbons (PAHs) — benzo[a]pyrene | Tobacco smoke, charbroiled food | Lung, oral cavity | Procarcinogen; CYP-activated to diol-epoxide; G→T transversions in TP53/KRAS |
| β-Naphthylamine, benzidine | Rubber/dye industry | Urinary bladder | Procarcinogen; hepatic acetylation detoxifies, but urinary deconjugation reactivates it at bladder urothelium |
| Aflatoxin B1 | Aspergillus flavus on peanuts/grain | Liver (HCC) | Procarcinogen; CYP3A4 → epoxide; G→T transversion at codon 249 of TP53 (hotspot mutation) |
| Nitrosamines | Cured/pickled foods; tobacco | Gastric, oesophageal | CYP-activated; prevalent in high-gastric-cancer regions |
| Asbestos | Mining, insulation | Mesothelioma, lung (co-carcinogen with tobacco) | Physical fibre mechanism + ROS; synergises with smoking for lung cancer (multiplicative risk) |
| Vinyl chloride | PVC industry | Angiosarcoma liver | Direct alkylation after CYP activation |
Note the tissue specificity of carcinogens — explained by local metabolic activation, target cell vulnerability, or topographic concentration (e.g., urothelium concentrates urinary carcinogens).