Page 8 of 24
PA6.2-3 | Molecular Basis of Cancer & Carcinogenesis — SDL Guide (Part 3)
Radiation Carcinogenesis
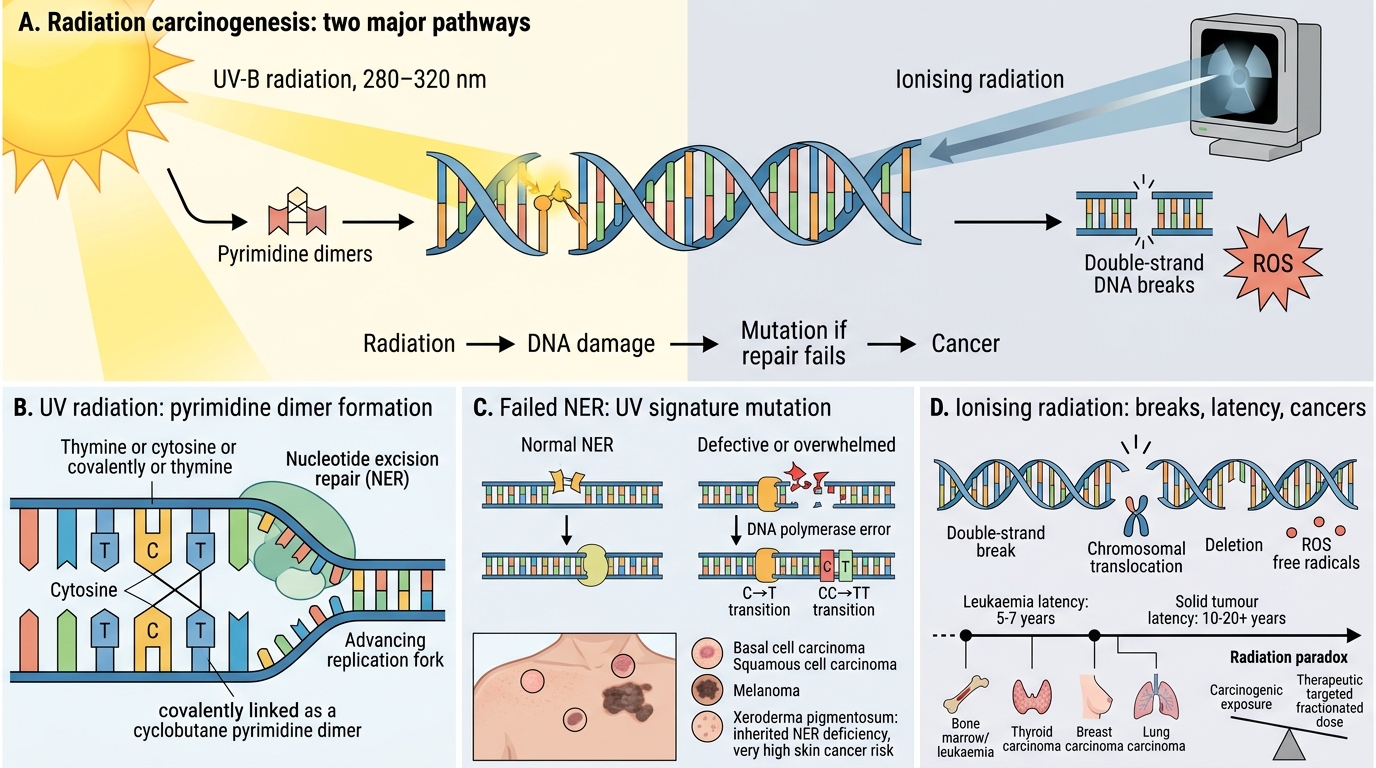
Radiation Carcinogenesis
Radiation damages DNA either directly or through reactive oxygen species (ROS).
Ultraviolet (UV) radiation:
• Source: sunlight (UV-B, 280–320 nm)
• Mechanism: adjacent pyrimidines on the same DNA strand form pyrimidine dimers (cyclobutane dimers), blocking replication. Normally removed by nucleotide excision repair (NER).
• Consequence: if NER is overwhelmed or defective, replication across the dimer causes C→T or CC→TT transitions — the UV signature mutation.
• Cancers: basal cell carcinoma, squamous cell carcinoma, melanoma (especially with intermittent intense exposure)
• Classic genetic disease: xeroderma pigmentosum — inherited NER deficiency; 1000× increased skin cancer risk
Ionising radiation (IR):
• Sources: X-rays, gamma rays, alpha/beta particles, radon gas
• Mechanism: double-strand DNA breaks, chromosomal translocations, deletions, and point mutations via ROS.
• Dose–response: roughly linear, no threshold — any dose carries some risk.
• Cancers: leukaemias (peak latency 5–7 years; dose-dependent — documented in Hiroshima/Nagasaki survivors), thyroid carcinoma (especially post-Chernobyl in children), breast, lung, sarcomas.
• Latency: solid tumours have longer latency (10–20+ years) than leukaemias.
• Paradox: ionising radiation is both carcinogenic and therapeutic — this is explained by the difference between fractionated targeted high-dose therapy (tumour cell killing) and low-dose scattered exposure (mutagenesis without lethal damage).
SELF-CHECK
A 7-year-old girl has freckling on sun-exposed skin, multiple skin cancers, and is found to have defective nucleotide excision repair. UV exposure leads to which PRIMARY DNA lesion that this pathway normally corrects?
A. Purine depurination
B. Double-strand breaks with chromosomal translocation
C. Cyclobutane pyrimidine dimers causing replication-blocking distortions
D. O6-methylguanine adducts from alkylating agents
Reveal Answer
Answer: C. Cyclobutane pyrimidine dimers causing replication-blocking distortions
UV-B radiation causes formation of cyclobutane pyrimidine dimers (and 6-4 photoproducts) between adjacent pyrimidines. Nucleotide excision repair (NER) recognises and removes these bulky adducts. Defective NER (as in xeroderma pigmentosum) leads to accumulation of these lesions, C→T transitions, and skin cancer. Double-strand breaks and alkylation adducts are caused by ionising radiation and alkylating agents, respectively.
Microbial and Viral Carcinogenesis
Approximately 15–20% of all human cancers worldwide are attributable to infectious agents. The major mechanisms are:
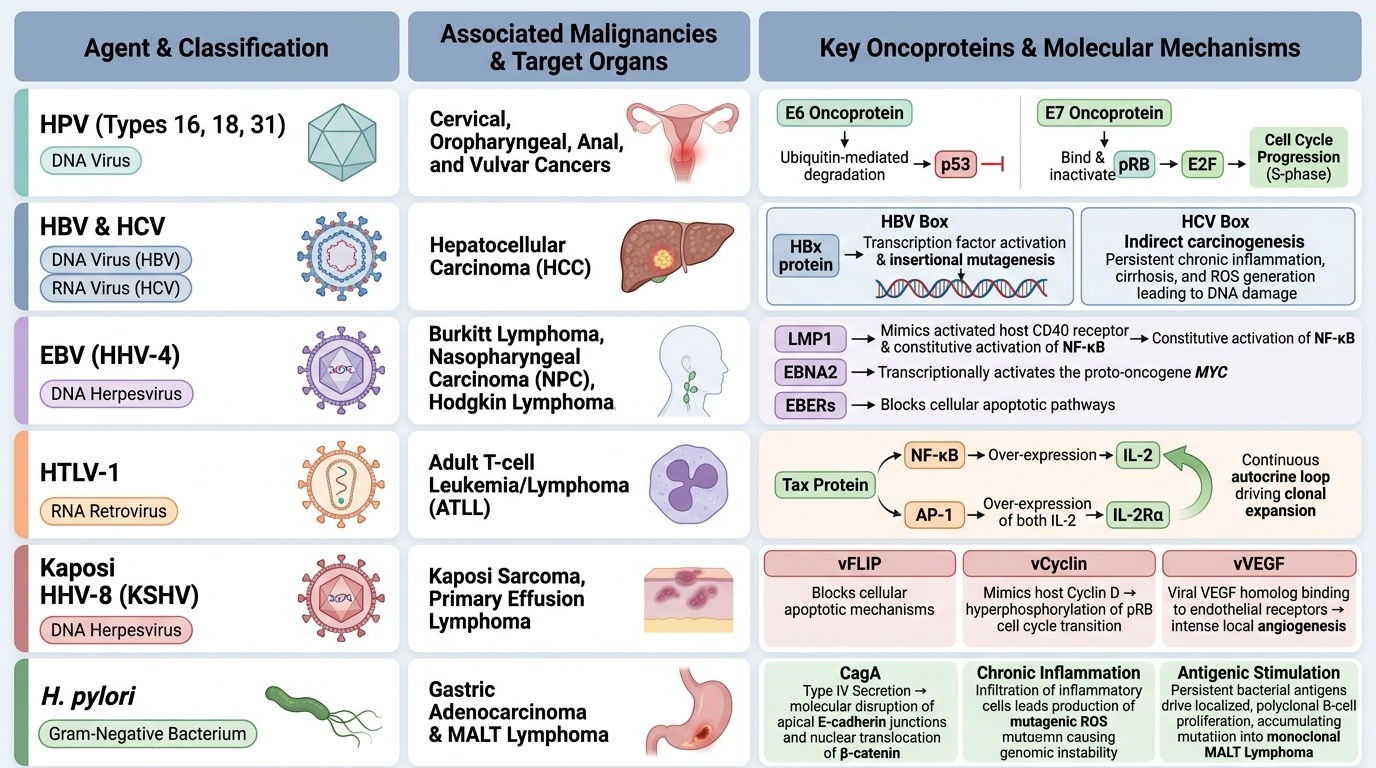
Provided image
- Integration of viral oncogenes — viral genome integrates and delivers oncoproteins that directly inactivate TSGs or activate growth signals
- Insertional mutagenesis — viral DNA integrates near a proto-oncogene, upregulating it
- Chronic inflammation — infection-driven cytokine milieu and ROS increase mutation rate and promote clonal expansion (H. pylori, HBV/HCV)
- Immunosuppression — allows pre-existing oncogenic co-infections to flourish (EBV in transplant recipients)
Key oncogenic agents:
| Agent | Type | Cancer | Key mechanism |
|---|---|---|---|
| HPV (16, 18, 31) | DNA virus | Cervical, oropharyngeal, anal, vulvar | E6 protein → ubiquitin-mediated degradation of p53; E7 → binds pRB, releasing E2F |
| HBV/HCV | DNA / RNA virus | Hepatocellular carcinoma (HCC) | HBV: HBx protein activates growth signals + insertional mutagenesis; HCV: indirect via cirrhosis/inflammation |
| EBV (HHV-4) | DNA herpesvirus | Burkitt lymphoma, NPC, Hodgkin lymphoma, PTLD | LMP1 mimics CD40 → NF-κB activation; EBNA2 activates MYC; EBERs suppress apoptosis |
| HTLV-1 | RNA retrovirus | Adult T-cell leukaemia/lymphoma (ATLL) | Tax protein activates NF-κB and AP-1; stimulates IL-2Rα (autocrine loop) |
| HHV-8 (KSHV) | DNA herpesvirus | Kaposi sarcoma, primary effusion lymphoma | vFLIP blocks apoptosis; vCyclin drives cycle; VEGF homologue drives angiogenesis |
| H. pylori | Bacterium | Gastric adenocarcinoma, MALT lymphoma | CagA oncoprotein disrupts E-cadherin; chronic inflammation + ROS mutagenesis; antigen-driven B-cell proliferation (MALT) |
CLINICAL PEARL
HPV vaccination as primary cancer prevention: HPV types 16 and 18 together cause ~70% of cervical cancers worldwide. The bivalent/quadrivalent/nonavalent vaccines train the immune system before infection. Understanding the E6/E7 mechanism (p53 and pRB degradation) explains why vaccination before sexual debut is so effective — it prevents the molecular first hit that initiates the multistep process. This is the most direct application of molecular carcinogenesis to public health.
SELF-CHECK
HPV type 16 E6 and E7 oncoproteins are found in a cervical biopsy. E7 exerts its carcinogenic effect PRIMARILY by:
A. Activating RAS-MAPK signalling through growth factor receptor mimicry
B. Binding pRB and releasing E2F transcription factor, driving uncontrolled S-phase entry
C. Inhibiting DNA mismatch repair, causing microsatellite instability
D. Upregulating BCL2, preventing apoptosis
Reveal Answer
Answer: B. Binding pRB and releasing E2F transcription factor, driving uncontrolled S-phase entry
HPV E7 binds and inactivates the retinoblastoma protein (pRB). Normally, pRB sequesters the E2F transcription factor; when pRB is phosphorylated (or in this case, bound by E7), E2F is released and drives transcription of genes needed for S-phase entry. E6 separately binds and targets p53 for proteasomal degradation. Together, E6 and E7 disable both major tumour suppressor pathways simultaneously.
Integrating the Molecular Framework
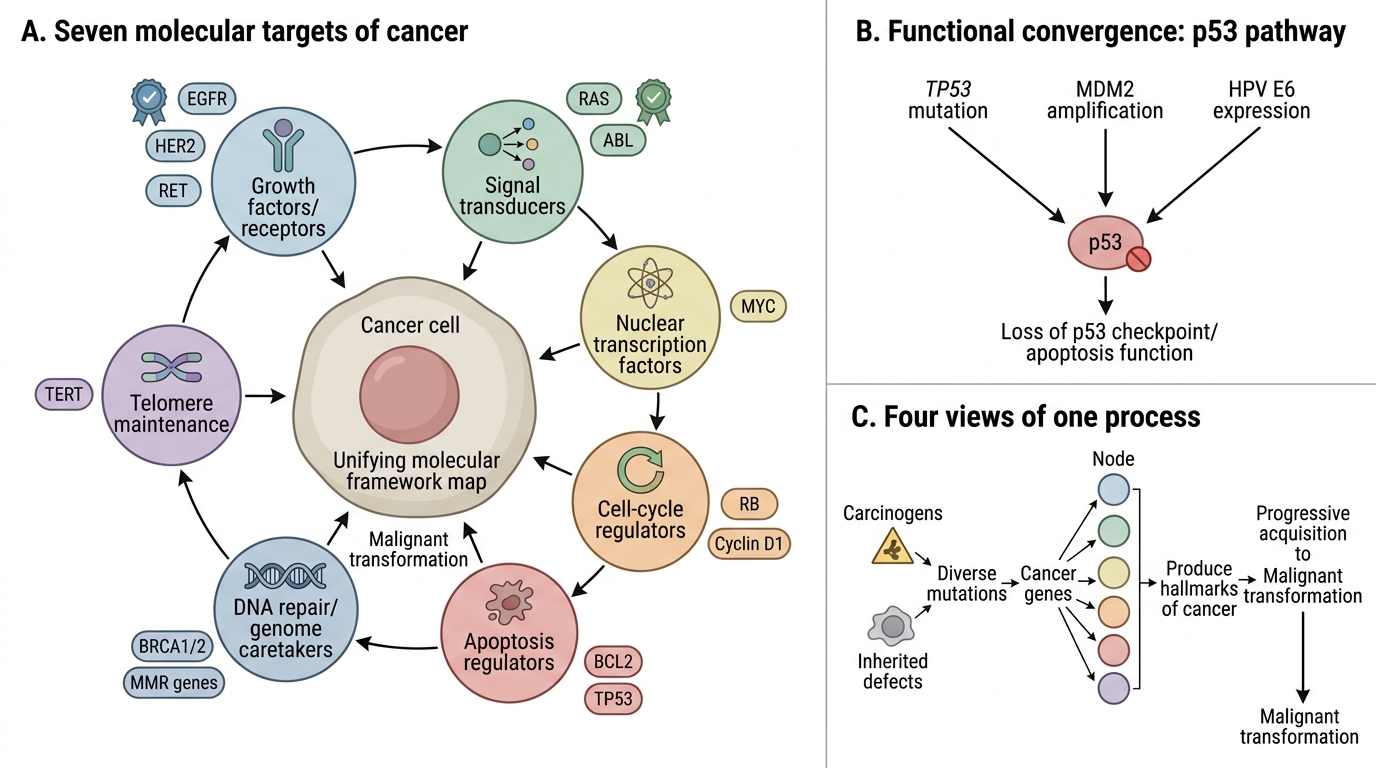
Integrated Molecular Framework of Cancer
It is useful to map all cancer genes onto a unifying framework:
The seven molecular targets of cancer:
- Growth factors/receptors — oncogenes (EGFR, HER2, RET)
- Signal transducers — oncogenes (RAS, ABL)
- Nuclear transcription factors — oncogenes (MYC)
- Cell-cycle regulators — TSGs (RB) and oncogenes (Cyclin D1)
- Apoptosis regulators — anti-apoptotic oncogenes (BCL2) and pro-apoptotic TSGs (TP53)
- DNA repair/genome caretakers — TSGs (BRCA1/2, MMR genes)
- Telomere maintenance — oncogenic activation of telomerase (TERT)
This map also explains why cancers are heterogeneous: many different mutations can hit the same functional node. For example, TP53 mutation, MDM2 amplification, or HPV E6 expression all disable p53 function — functionally convergent, molecularly distinct.
The hallmarks, cancer genes, multistep model, and carcinogens are not four separate topics but four views of the same phenomenon: a cell progressively acquiring, through diverse mechanisms, the traits that allow it to escape normal constraints and become malignant.