Page 13 of 22
PA9.1-2 | Amyloidosis — SDL Guide
Learning Objectives
- Define amyloid and describe its physical and chemical nature including the β-pleated sheet configuration
- Explain the pathogenesis of amyloid deposition — protein misfolding and impaired degradation
- Classify amyloidosis by precursor protein: AL, AA, Aβ2-microglobulin, ATTR, Aβ, and endocrine forms
- Distinguish systemic from localised amyloidosis and identify the clinical conditions associated with each type
- Describe the gross and microscopic morphology of amyloid deposits, including the Congo red staining reaction
- Enumerate the major organs affected in systemic amyloidosis and the clinical consequences of each
INSTRUCTIONS
Amyloidosis is a unifying concept in pathology — a single abnormal physical property (the β-pleated sheet) underlies diverse diseases ranging from plasma cell dyscrasia to Alzheimer disease. Mastering the classification logic and the diagnostic Congo red reaction will pay dividends in every system you study from Year 2 onward. This module links directly to haematology (AL type), chronic inflammation (AA type), and neuroscience (Aβ), and will recur in clinical postings whenever a patient presents with unexplained heavy proteinuria or restrictive cardiac disease.
References
- Robbins & Kumar: Basic Pathology, 11th ed., Ch 4 (Cellular Responses to Stress) and Ch 11 (Diseases of Immunity) (textbook)
- Harsh Mohan: Textbook of Pathology, 8th ed., Ch 4 (Amyloidosis) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 58-year-old man with a 10-year history of rheumatoid arthritis presents with 4+ pitting oedema, frothy urine, and serum albumin of 1.8 g/dL. His renal biopsy shows amorphous eosinophilic material in the glomerular mesangium that stains salmon-pink with Congo red — and glows apple-green under polarised light.
A different patient, 65 years old, with a known plasma cell myeloma develops the same renal picture, plus macroglossia and carpal tunnel syndrome.
Same end-organ damage. Very different protein culprits. By the end of this module you will know exactly why — and how a single histochemical stain unifies both diagnoses.
WHY THIS MATTERS
Amyloidosis is not a single disease but a shared structural endpoint reached by more than 30 different proteins. Understanding it at the protein-folding level explains:
- Why chronic inflammatory diseases (RA, TB, IBD) destroy the kidney over decades
- Why myeloma kills through renal failure as often as marrow failure
- Why dialysis patients develop carpal tunnel syndrome
- Why Alzheimer disease and type 2 diabetes share a structural motif with systemic amyloidosis
For the clinician, recognising amyloidosis early — before end-organ failure — requires knowing which patients are at risk and which biopsy site gives the highest yield.
RECALL
Before continuing, briefly recall:
- What is a β-pleated sheet secondary protein structure, and how does it differ from an α-helix?
- What are immunoglobulin light chains (κ and λ)? Which cell type overproduces them in plasma cell dyscrasia? (cross-reference: Cluster H11 — Plasma Cell Neoplasms)
- What is the acute-phase response, and which liver-derived proteins rise during chronic inflammation?
- What does Congo red normally stain, and what optical property is needed to detect birefringence?
If any of these are unclear, spend 3 minutes reviewing before proceeding — this module builds directly on all four concepts.
What Is Amyloid? Physical and Chemical Nature
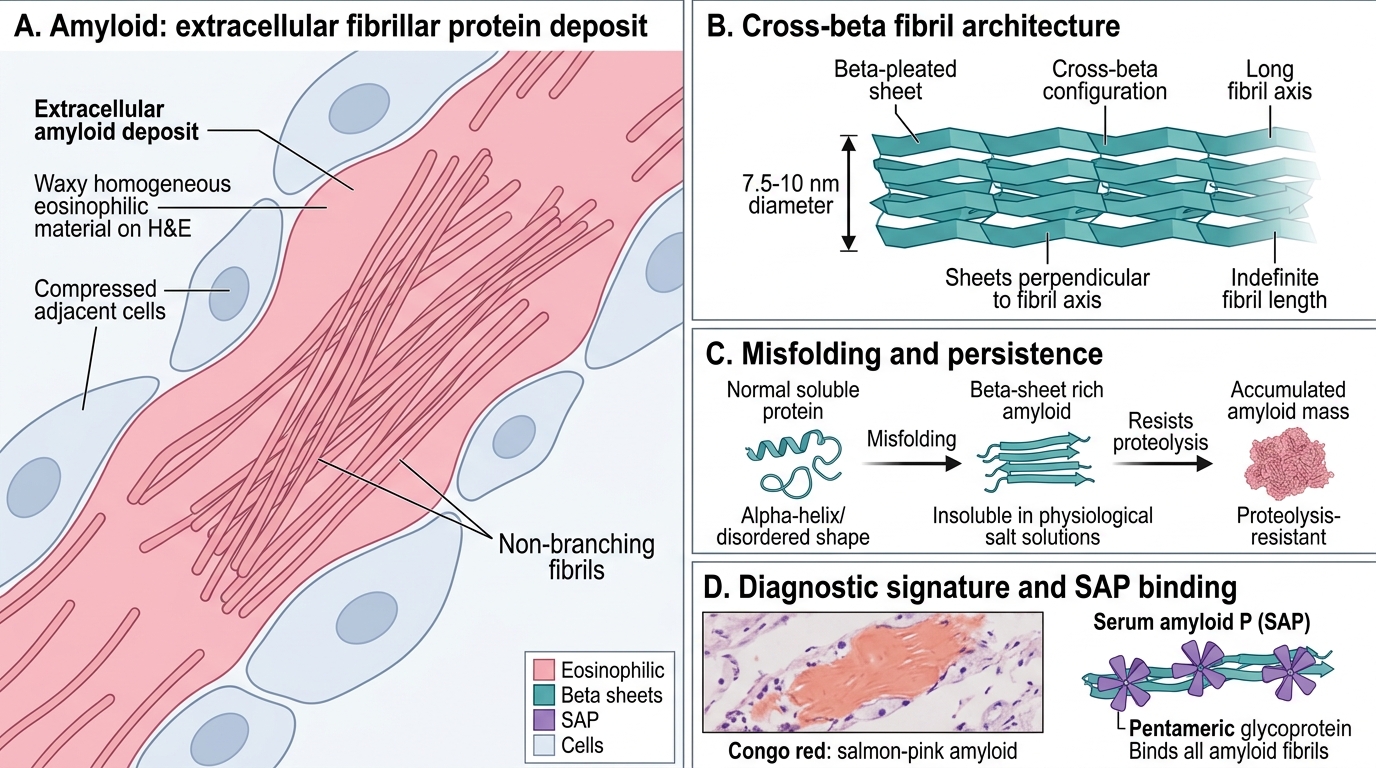
Amyloid is a pathological extracellular deposit of insoluble fibrillar proteins that share a unique secondary structure regardless of the precursor protein involved.
Key physical and chemical properties:
| Property | Detail |
|---|---|
| Structure | β-pleated sheet (cross-β) configuration — sheets stacked perpendicular to the fibril axis |
| Solubility | Insoluble in physiological salt solutions; resists proteolysis |
| Fibrils | 7.5–10 nm diameter, non-branching, indefinite length |
| P component | Serum amyloid P (SAP) — a pentameric glycoprotein that binds all amyloid fibrils (used in scintigraphy) |
| Appearance | Waxy, homogeneous, eosinophilic on H&E |
The β-pleated sheet is the unifying feature. Proteins that normally have α-helical or disordered conformations can misfold into this configuration under the right conditions — and once they do, they resist degradation and accumulate indefinitely.
Staining — the diagnostic signature:
Amyloid stains salmon-pink with Congo red on standard light microscopy. Under polarised light, the same section exhibits apple-green birefringence — this optical property is pathognomonic for amyloid and is caused by the parallel alignment of dye molecules along the β-pleated sheet.
Other stains: crystal violet (metachromatic purple), thioflavin T (fluorescence) — used in research.
Iodine reaction: amyloid stains brown with Lugol's iodine and turns violet-blue with dilute H₂SO₄ — historically used at autopsy (the "lardaceous" or "waxy" appearance that early pathologists described).
Amyloid: Physical and Chemical Nature
SELF-CHECK
A renal biopsy shows amorphous eosinophilic extracellular deposits. Under polarised light after Congo red staining, the deposits show apple-green birefringence. What structural property of amyloid is responsible for this optical phenomenon?
A. α-helical coiling of the fibril proteins
B. Parallel alignment of Congo red molecules along β-pleated sheets
C. Glycosylation of the serum amyloid P component
D. Cross-linking of collagen fibrils by transglutaminase
Reveal Answer
Answer: B. Parallel alignment of Congo red molecules along β-pleated sheets
The β-pleated sheet configuration forces Congo red molecules to align in a parallel, ordered array along the fibril axis. This ordered arrangement of dye molecules acts as a diffraction grating under polarised light, producing the characteristic apple-green birefringence. This is NOT a property of collagen, α-helices, or the P component.
Pathogenesis — Misfolding and Impaired Degradation
Amyloid Pathogenesis: Misfolding, Impaired Degradation, and Seeding
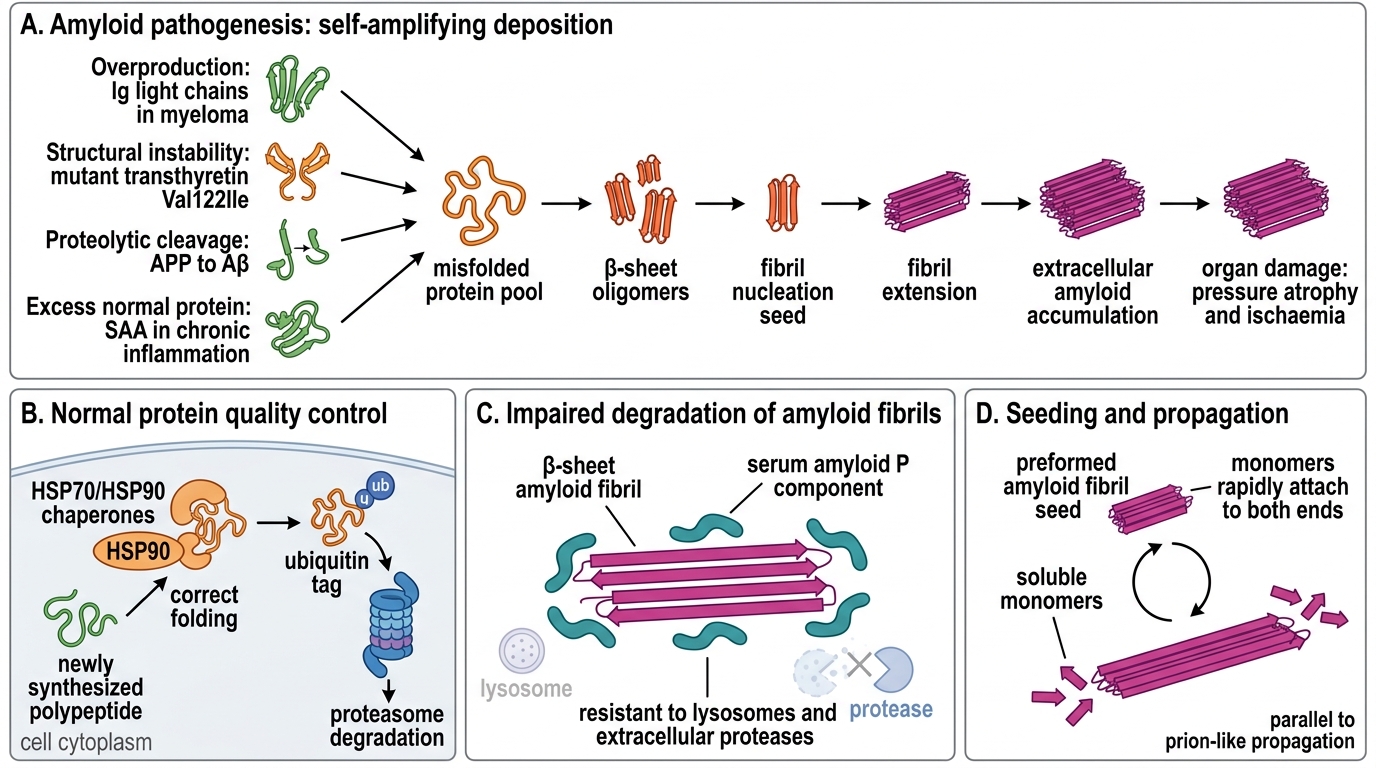
Amyloid accumulation results from two converging mechanisms:
1. Protein misfolding (aggregation-prone precursors)
Normally, chaperone proteins (HSP70, HSP90) assist correct folding, and the ubiquitin-proteasome system degrades misfolded proteins. Amyloidogenic proteins escape this surveillance for one or more reasons:
- Overproduction overwhelms clearance — immunoglobulin light chains in myeloma
- Structural instability — mutant transthyretin (Val122Ile) misfolds spontaneously
- Proteolytic cleavage generates a fragment that is aggregation-prone — APP → Aβ in Alzheimer
- Normal-sequence protein released in excess — SAA in chronic inflammation
2. Impaired degradation
Once β-sheet oligomers and fibrils form, they are highly resistant to lysosomes and extracellular proteases. The serum amyloid P component actually protects fibrils from proteolysis, prolonging their survival.
Key concept — seeding: Preformed amyloid fibrils act as nucleation seeds, accelerating the addition of new monomers. This is why amyloid deposition is self-amplifying once initiated — a parallel to prion propagation.
Summary pathway:
Precursor protein overproduction or structural instability → misfolding → β-sheet oligomers → fibril nucleation (seeding) → fibril extension → extracellular accumulation → organ damage via pressure atrophy and ischaemia
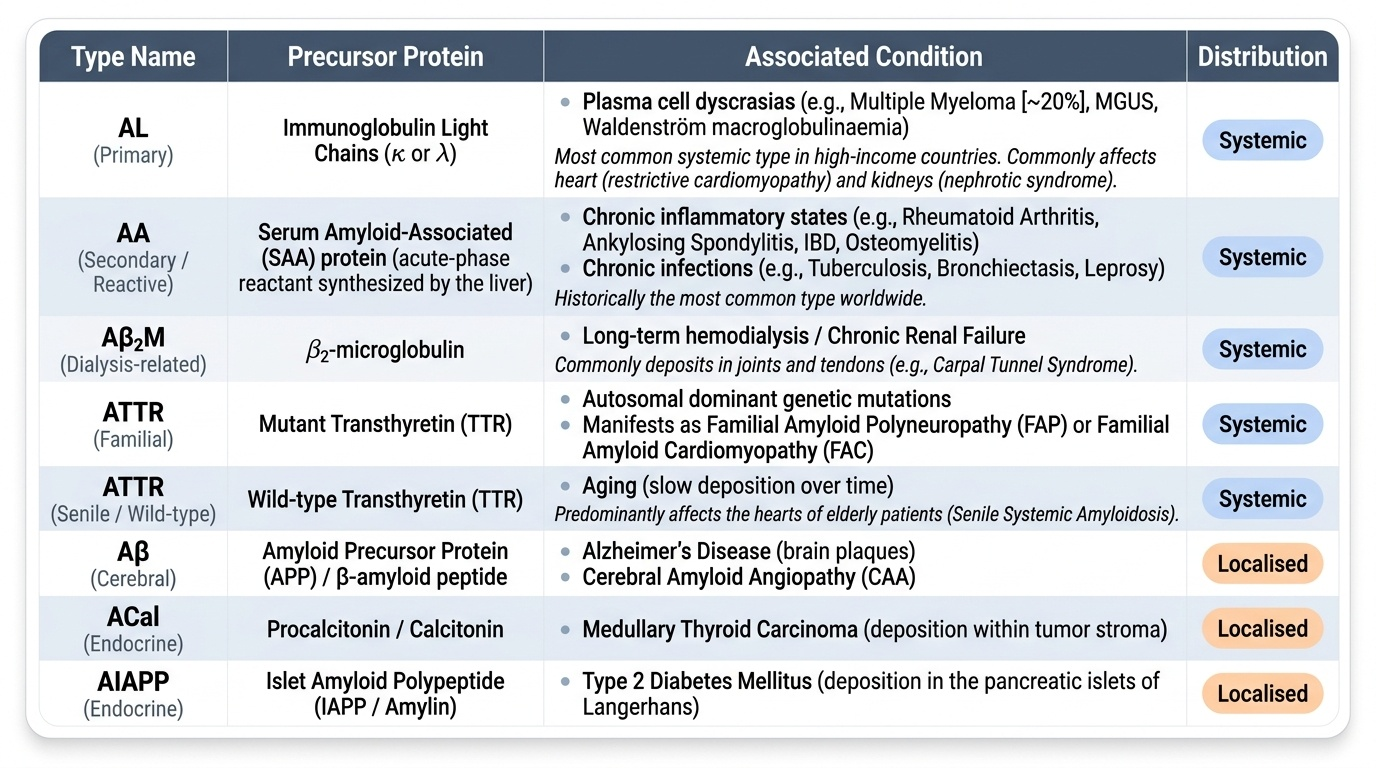
Classification by Precursor Protein
Amyloidosis is classified by the precursor protein that forms the fibril. The letter designation follows: protein abbreviation + 'A' prefix (e.g., AL = Amyloid of Light chain origin).
Provided image
Systemic amyloidosis (generalised):
AL (Primary amyloidosis)
• Precursor: Immunoglobulin light chains (κ > λ, or λ > κ depending on clone)
• Association: Plasma cell dyscrasia — multiple myeloma (20%), monoclonal gammopathy (MGUS), Waldenström macroglobulinaemia
• Cross-reference: Cluster H11 (Plasma Cell Neoplasms)
• Organs affected: heart (restrictive), kidney (nephrotic), peripheral nerves, tongue (macroglossia), carpal tunnel
• Note: AL is the most common type of systemic amyloidosis in high-income countries
AA (Secondary / Reactive systemic amyloidosis)
• Precursor: Serum amyloid-associated (SAA) protein — an acute-phase reactant synthesised by the liver
• Association: Chronic inflammatory states — rheumatoid arthritis, ankylosing spondylitis, tuberculosis, bronchiectasis, chronic osteomyelitis, inflammatory bowel disease (Crohn > UC), leprosy
• Historically the commonest type worldwide (TB belt)
• Organs preferentially affected: kidney (most common and most devastating), liver, spleen, adrenals
Aβ2-microglobulin amyloidosis (Aβ2M)
• Precursor: β2-microglobulin (MHC class I light chain) — NOT cleared by dialysis membranes
• Association: Long-term haemodialysis (>10 years)
• Clinically: carpal tunnel syndrome, destructive arthropathy
ATTR (Transthyretin amyloidosis)
• Precursor: Transthyretin (formerly prealbumin) — a transport protein for thyroxine and retinol
• Familial (hereditary) ATTR: point mutations (Val30Met most common); autosomal dominant; peripheral neuropathy + cardiac involvement
• Senile/wild-type ATTR: normal-sequence transthyretin deposits in elderly men; restrictive cardiomyopathy is the dominant feature
Localised amyloidosis:
Aβ (Alzheimer-type)
• Precursor: Amyloid-β peptide — a 40–42 amino acid fragment of amyloid precursor protein (APP)
• Site: Brain — senile plaques (Aβ core) and amyloid angiopathy
• Down syndrome (trisomy 21): extra APP gene copy → early Alzheimer deposits by age 40
Endocrine amyloid (localised)
• Medullary thyroid carcinoma: calcitonin-derived amyloid in tumour stroma
• Islet amyloid (IAPP): amylin (islet amyloid polypeptide) in type 2 diabetes mellitus — deposits in islets of Langerhans
CLINICAL PEARL
AL vs AA — the clinical differentiator: In AL amyloidosis, look for monoclonal protein in serum/urine (SPEP, UPEP, free light chain assay) and a bone marrow biopsy showing clonal plasma cells. In AA amyloidosis, look for an obvious chronic inflammatory disease in the history. Tissue typing via mass spectrometry or immunohistochemistry (anti-κ/λ, anti-SAA) is increasingly used to type amyloid in biopsy specimens because immunohistochemistry on formalin-fixed paraffin-embedded tissue is notoriously unreliable alone.
Dialysis amyloid clue: A dialysis patient with bilateral carpal tunnel syndrome, especially after >10 years on haemodialysis, almost always has Aβ2M amyloidosis until proven otherwise.
SELF-CHECK
A 45-year-old woman with a 15-year history of rheumatoid arthritis develops progressive nephrotic syndrome. Renal biopsy shows Congo red-positive deposits. Which precursor protein is most likely responsible?
A. Immunoglobulin light chain (AL)
B. Transthyretin (ATTR)
C. Serum amyloid-associated protein (AA)
D. β2-microglobulin (Aβ2M)
Reveal Answer
Answer: C. Serum amyloid-associated protein (AA)
Chronic active rheumatoid arthritis drives sustained elevation of the acute-phase reactant SAA. Persistent elevation over years leads to AA amyloidosis, which preferentially deposits in the kidney, causing nephrotic syndrome and eventually renal failure. AL (option A) would require evidence of a plasma cell clone; ATTR (B) causes cardiomyopathy in the elderly; Aβ2M (D) requires long-term dialysis.