Page 5 of 20
PA11.1-3 | Genetic & Pediatric Diseases — SDL Guide
Learning Objectives
- Classify genetic mutations (point, frameshift, trinucleotide repeat) and identify their inheritance patterns (AD, AR, X-linked, mitochondrial).
- Describe the pathogenesis and clinico-pathological features of major cytogenetic abnormalities — trisomy 21, trisomy 18, trisomy 13, Turner syndrome (45,X), and Klinefelter syndrome (47,XXY).
- Recognise the small round blue cell tumours of childhood — neuroblastoma, Wilms tumour, retinoblastoma, Ewing sarcoma, and rhabdomyosarcoma — and state their key molecular markers.
- Explain the pathogenesis of lysosomal storage disorders and describe the hallmark morphology of Gaucher, Niemann-Pick, Tay-Sachs, mucopolysaccharidoses, and glycogen storage diseases.
INSTRUCTIONS
Genetic and pediatric diseases account for a disproportionate share of childhood mortality in India and are tested heavily in MBBS Part-II theory and USMLE-style integrated exams. Understanding these conditions builds directly on your Year-1 biochemistry (enzyme kinetics, DNA replication) and embryology, and is essential for interpreting paediatric case presentations in clinical years. This module uses a mechanism-first approach: grasp the molecular defect → predict the phenotype → recognise the pathology slide.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch 5 (Genetic Disorders) (textbook)
- Harsh Mohan Textbook of Pathology, 8th ed., Ch 8 & 9 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 6-month-old girl is brought in with hypotonia, feeding difficulty, upslanting palpebral fissures, a single palmar crease, and a ventricular septal defect. Her karyotype returns as 47,XX+21. Meanwhile, in the next cubicle, a 3-year-old boy has a palpable abdominal mass — ultrasound shows a heterogeneous renal tumour. And in the genetics clinic, a 2-year-old presents with progressive cherry-red macular spots and developmental regression. Three children, three entirely different mechanisms, but all traceable to a single principle: the genome, when altered in sequence, number, or enzyme function, derails normal development in characteristic, predictable ways.
WHY THIS MATTERS
Genetic disorders are the leading cause of infant mortality in developed countries and account for a substantial — and growing — fraction of paediatric admissions in India as infectious disease burden falls. As a clinician you will encounter Down syndrome in every posting, storage disorders in specialty clinics, and paediatric tumours in general surgery and oncology. The NMC CBUC 2024 curriculum places PA11.1–11.3 as must-know competencies, and these topics appear in both viva and OSPE stations.
RECALL
Before proceeding, activate your Year-1 knowledge:
- DNA replication errors: substitution, deletion, insertion — and how reading frames are affected.
- Mendelian inheritance: dominant vs. recessive; X-linked hemizygosity in males.
- Cell division: mitosis vs. meiosis; non-disjunction as a source of aneuploid gametes.
- Lysosome function: acid hydrolases digest macromolecules; substrate accumulates when an enzyme is absent.
- Tumour suppressor genes (TSG): loss of heterozygosity (LOH); the two-hit hypothesis (Knudson).
If any of these feel shaky, spend 5 minutes on your Year-1 biochemistry notes before continuing — these concepts are the engine for everything that follows.
Types of Genetic Mutations & Inheritance Patterns
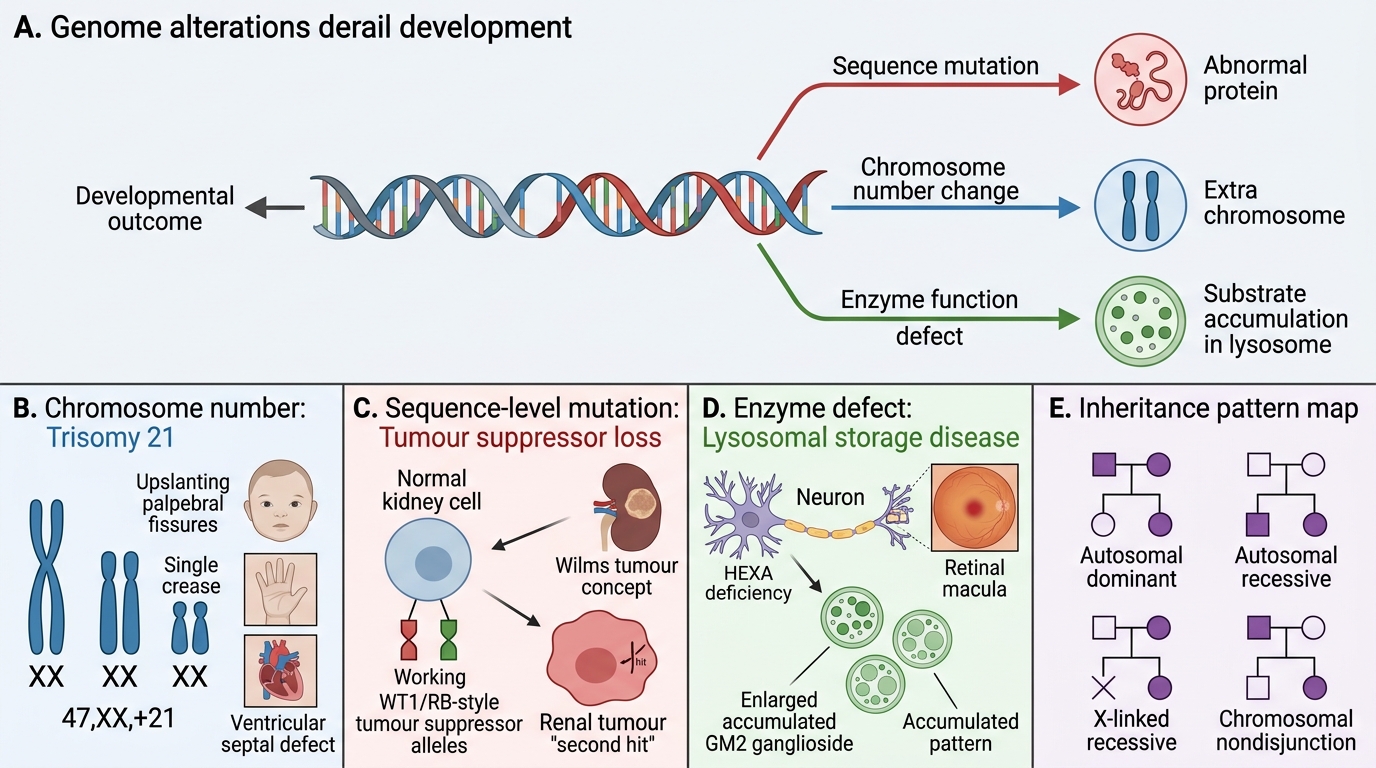
Genetic Alterations and Clinical Patterns
Genetic disease arises from alterations at three levels: single nucleotide, reading frame, or whole chromosome.
Point mutations substitute one base for another. A missense mutation changes one amino acid (e.g., sickle-cell: glutamate→valine in β-globin). A nonsense mutation inserts a premature stop codon, truncating the protein.
Frameshift mutations result from insertions or deletions of nucleotides not divisible by three. Every codon downstream shifts, producing a garbled and usually non-functional protein. Even a single-base deletion can be catastrophic.
Trinucleotide repeat expansions are a special category: a short DNA motif (e.g., CAG, CGG) is repeated in tandem; beyond a threshold copy number the gene is silenced or produces a toxic protein. Huntington disease (CAG in HTT), Fragile-X (CGG in FMR1), and myotonic dystrophy follow this pattern.
Inheritance patterns — quick reference:
| Pattern | Key feature | Example |
|---|---|---|
| Autosomal dominant (AD) | One mutant allele sufficient; vertical transmission; 50% offspring risk | Huntington, Marfan, NF1 |
| Autosomal recessive (AR) | Two mutant alleles needed; horizontal (sibling) pattern; 25% risk | CF, PKU, most storage disorders |
| X-linked recessive | Carrier females, affected males; no male-to-male transmission | Haemophilia, G6PD deficiency |
| X-linked dominant | Affected males + carrier females; lethal in hemizygous males | Rett syndrome |
| Mitochondrial | Maternal transmission; affects all children of an affected mother; heteroplasmy | MELAS, Leber's optic neuropathy |
Numerical Cytogenetic Abnormalities — Autosomes
Aneuploidy results from non-disjunction during meiosis I or II, producing gametes with an extra or missing chromosome.
Trisomy 21 — Down syndrome (47,XX/XY+21)
The most common viable autosomal trisomy. Risk rises steeply with maternal age (1:1,500 at age 20 → 1:25 at age 45) because non-disjunction occurs predominantly in maternal meiosis I. 95% are free trisomy; 4% are Robertsonian translocations (chromosome 21 fused to 14 — FAMILIAL, not age-related); 1% mosaic.
Phenotype: intellectual disability, hypotonia, flat occiput, upslanting palpebral fissures, epicanthal folds, Brushfield spots, single palmar crease, wide sandal gap, short stature, macroglossia.
Pathology: endocardial cushion defects (ASD + VSD) in 40%; duodenal atresia ("double bubble" sign); Hirschsprung disease; acute megakaryoblastic leukaemia (AMKL); near-universal Alzheimer-type dementia by age 40 (APP gene on chromosome 21 → amyloid overproduction).
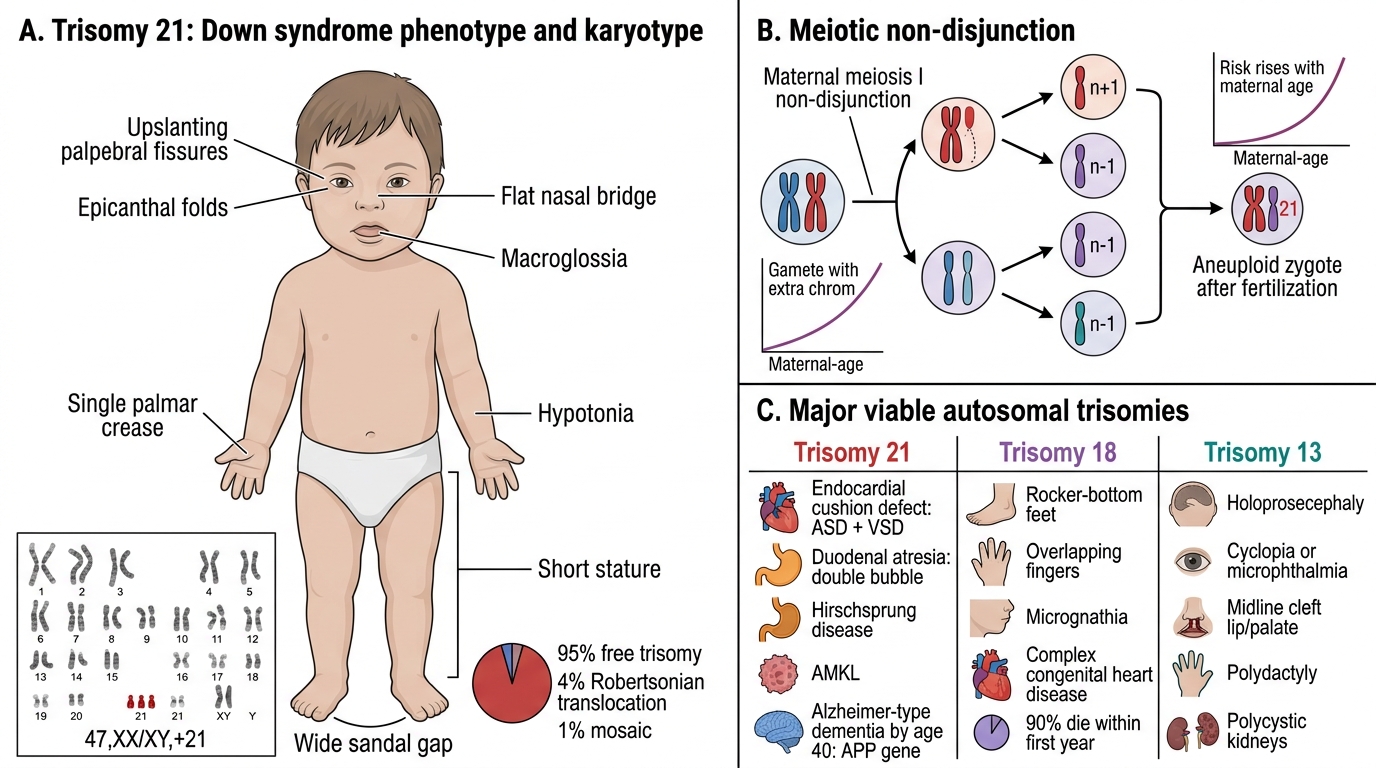
Autosomal Aneuploidies: Trisomy 21, 18, and 13
Trisomy 18 — Edwards syndrome: 3× lethal; features include rocker-bottom feet, overlapping fingers, micrognathia, complex congenital heart disease. 90% die within first year.
Trisomy 13 — Patau syndrome: holoprosencephaly, cyclopia/microphthalmia, midline cleft lip/palate, polydactyly, polycystic kidneys. Most lethal within weeks.
SELF-CHECK
A 2-month-old has upslanting palpebral fissures, a VSD, and hypotonia. Karyotyping shows 47+21. What is the most likely mechanism of non-disjunction in the majority of Down syndrome cases?
A. Paternal meiosis II error
B. Maternal meiosis I error
C. Robertsonian translocation
D. Somatic mitotic non-disjunction
Reveal Answer
Answer: B. Maternal meiosis I error
~95% of free trisomy 21 arises from non-disjunction during maternal meiosis I, explaining the strong maternal-age effect. Robertsonian translocations account for ~4% and are familial. Somatic mitotic non-disjunction produces the mosaic form (~1%).
Sex Chromosome Abnormalities — Turner & Klinefelter
Turner syndrome (45,X)
The only viable monosomy in humans. 99% of 45,X conceptions abort spontaneously — those that survive often carry mosaicism (45,X / 46,XX) which ameliorates severity. Affects ~1:2,500 live female births.
Key features: short stature (absent pubertal growth spurt), primary amenorrhoea, streak gonads (bilateral fibrous bands replacing ovaries → hypergonadrotropic hypogonadism), low posterior hairline, webbed neck (pterygium colli), shield chest, wide-spaced nipples, cubitus valgus, coarctation of the aorta (15%), horseshoe kidney.
Pathology: streak gonads carry risk of gonadoblastoma if Y-chromosomal material present; premature osteoporosis; absence of secondary sexual characteristics without oestrogen replacement.
⚑ AI image — pending faculty review (auto-QA score 6/10; best of 3 attempts)
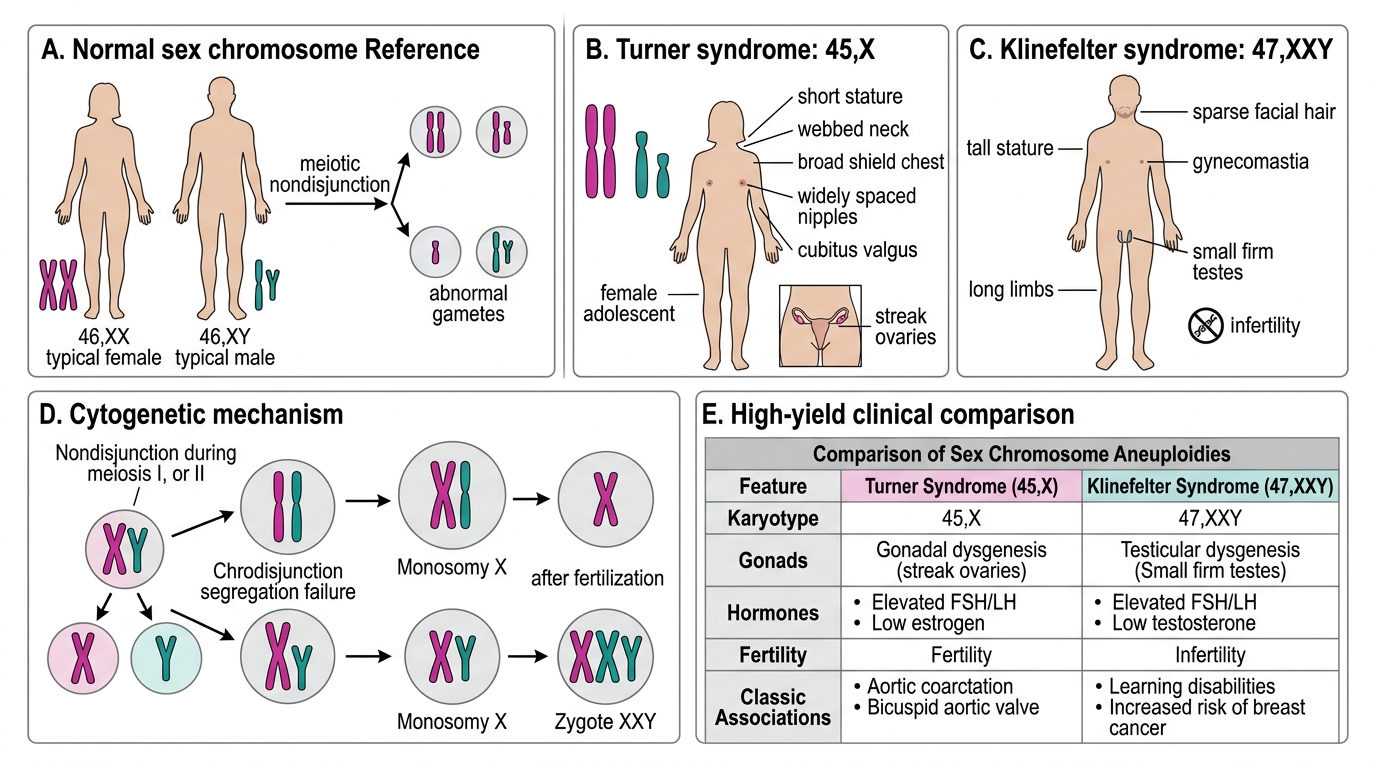
Sex Chromosome Abnormalities: Turner and Klinefelter Syndromes
Klinefelter syndrome (47,XXY)
Most common sex chromosome disorder in males (1:500–1,000). Extra X from either maternal (most common) or paternal non-disjunction.
Key features: tall stature with long legs, small firm testes (hyalinisation of seminiferous tubules → azoospermia), gynaecomastia, decreased facial/body hair, mild learning difficulties. Primary cause of male infertility via azoospermia.
Pathology on biopsy: hyalinised seminiferous tubules, Leydig cell hyperplasia, absence of spermatogonia → elevated FSH/LH, low testosterone → treatment with testosterone.
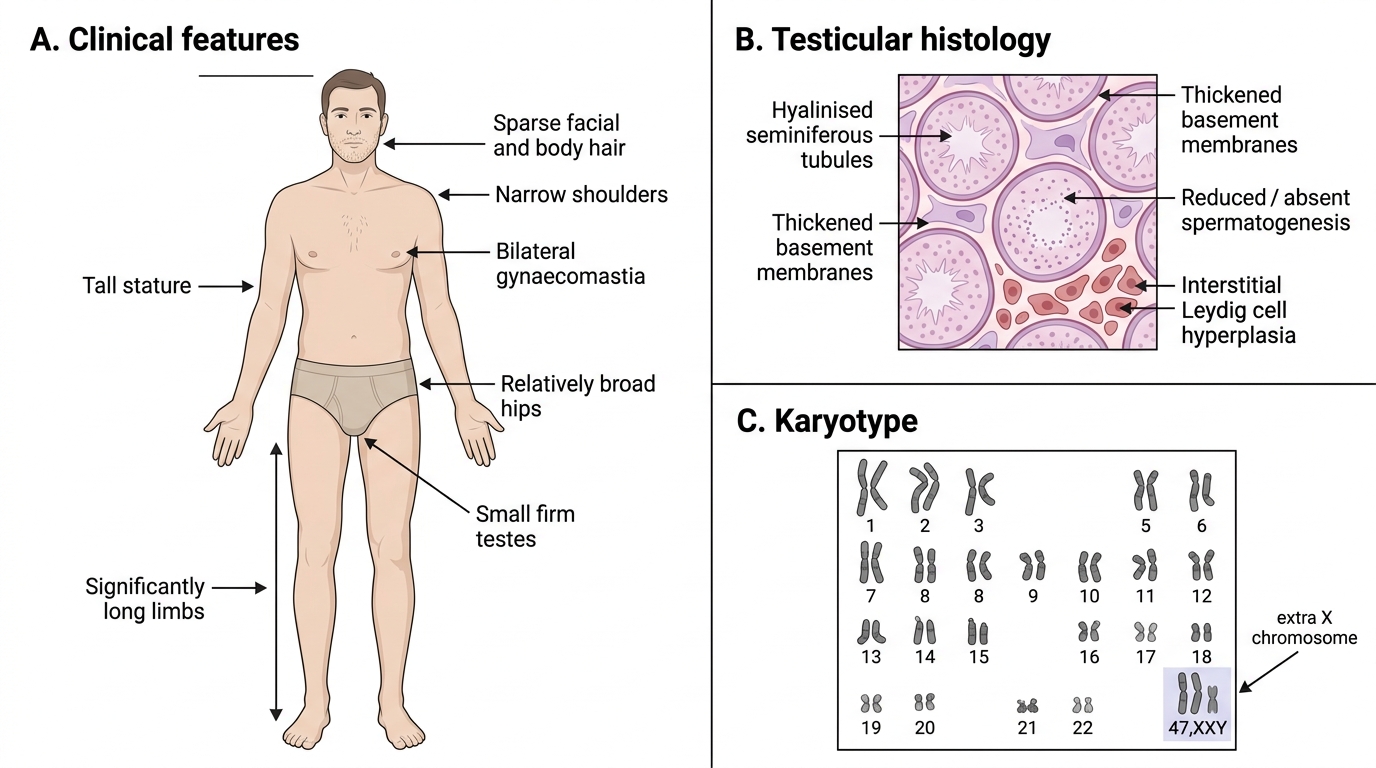
Klinefelter Syndrome: Clinical Features, Histology, and Karyotype