Page 6 of 20
PA11.1-3 | Genetic & Pediatric Diseases — SDL Guide (Part 2)
Structural Cytogenetic Abnormalities & Karyotype/FISH
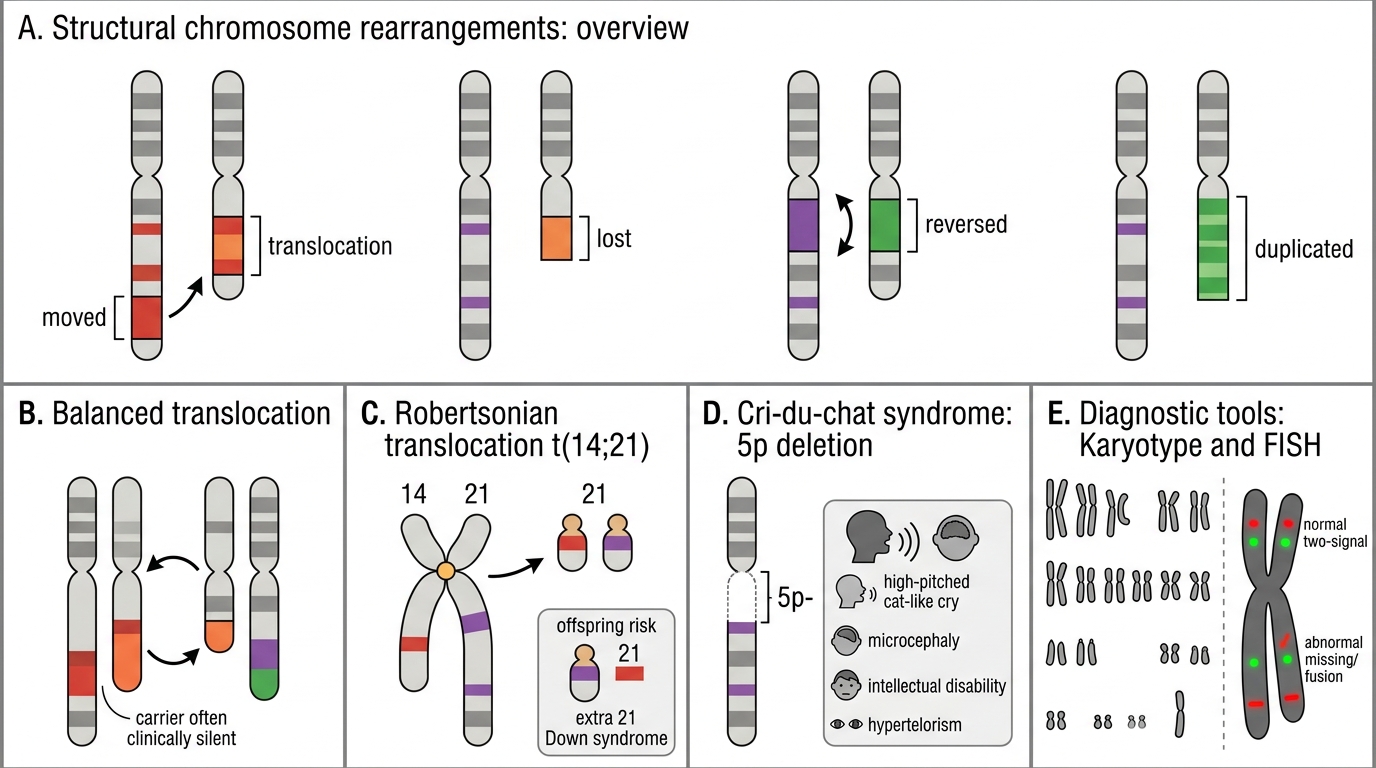
Structural Cytogenetic Abnormalities and Diagnostic Tests
Structural rearrangements arise from chromosome breakage and abnormal repair:
- Translocation: segment moves from one chromosome to another. Balanced translocations carry no net gain/loss — often clinically silent in the carrier but risk unbalanced offspring. Robertsonian translocation: two acrocentric chromosomes (13, 14, 15, 21, 22) fuse at centromere; a carrier of t(14;21) risks Down syndrome children.
- Deletion: loss of a chromosomal segment. Cri-du-chat syndrome: deletion of short arm of chromosome 5 (5p-); high-pitched cat-like cry (laryngeal hypoplasia), microcephaly, intellectual disability, hypertelorism.
- Inversion: segment reverses orientation. Pericentric (includes centromere) vs. paracentric.
- Duplication: extra copy of a segment; often better tolerated than deletion.
Diagnostic tools:
| Tool | Resolution | Best for |
|---|---|---|
| Karyotype (G-banding, 400–550 bands) | ~5–10 Mb | Numerical abnormalities, large deletions, translocations |
| FISH (Fluorescence In Situ Hybridisation) | ~100 kb | Targeted microdeletion (DiGeorge 22q11), confirmation of translocation |
| Array CGH / SNP array | 5–50 kb | Genome-wide copy number variants (CNVs) |
| PCR/sequencing | Single nucleotide | Specific point mutations (BRCA, TP53) |
Exam tip: FISH is ordered when you suspect a microdeletion too small for karyotype but too large for routine sequencing.
Paediatric Tumours — Hamartoma, Choristoma & Overview
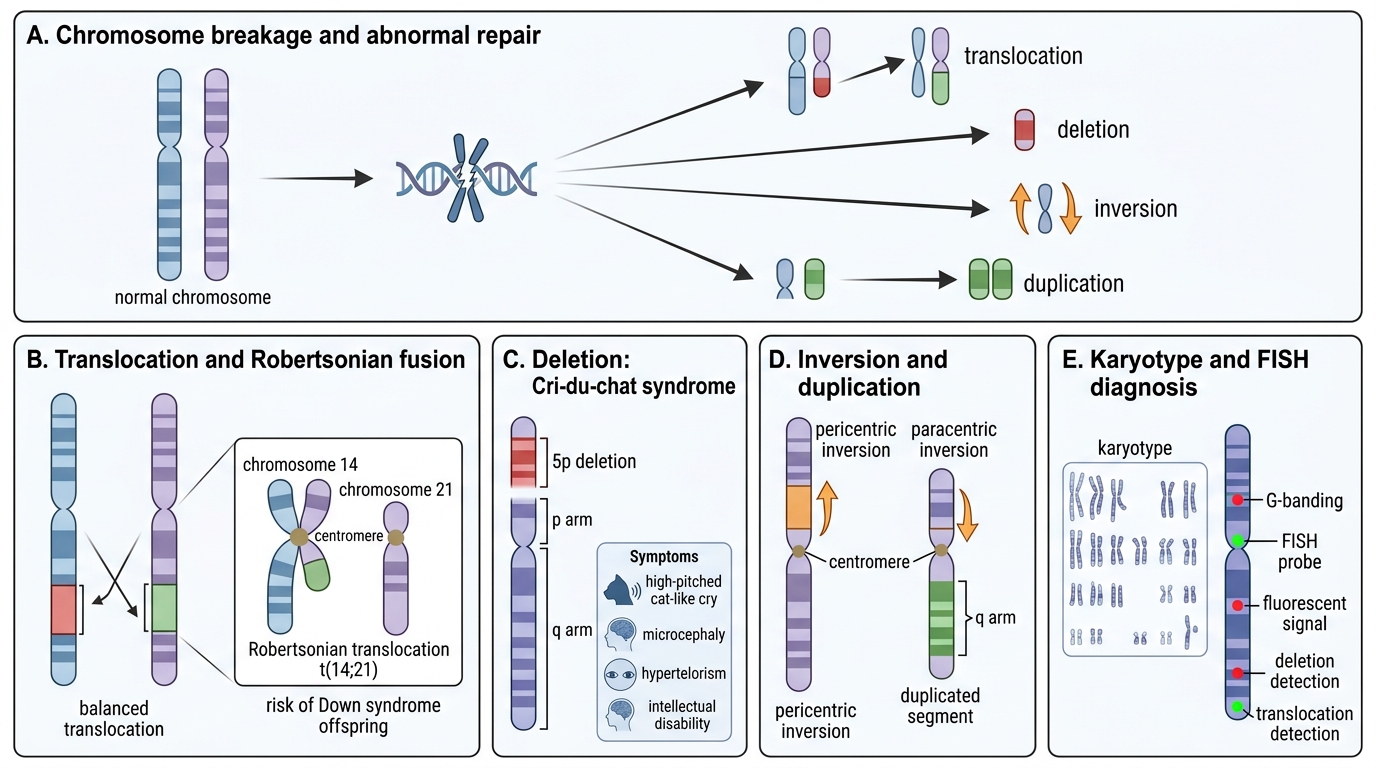
Structural Cytogenetic Abnormalities and Diagnostic Tools
Before examining specific tumours, two important developmental lesions:
Hamartoma: disorganised but normal tissue elements native to that site (e.g., pulmonary hamartoma with cartilage + epithelium + fat). Not a true neoplasm. Grows slowly, proportionate to body growth, does not metastasise.
Choristoma: normal tissue in an abnormal location (e.g., gastric mucosa in a Meckel's diverticulum causing peptic ulceration). Also called heterotopia.
The dominant paediatric malignancies are the small round blue cell tumours (SRBCT) — a group named for their shared histological appearance: sheets of small cells with scant cytoplasm and round hyperchromatic nuclei. The challenge — and the exam test — is distinguishing them using ancillary features.
| Tumour | Age | Site | Key marker |
|---|---|---|---|
| Neuroblastoma | <5 yr | Adrenal/sympathetic chain | N-myc amplification, catecholamines |
| Wilms (nephroblastoma) | 2–5 yr | Kidney | WT1 gene, triphasic histology |
| Retinoblastoma | <5 yr | Retina | RB1 gene, two-hit hypothesis |
| Ewing sarcoma | 5–20 yr | Bone (diaphysis) | t(11;22), EWSR1-FLI1 fusion |
| Rhabdomyosarcoma | <10 yr (embryonal); teen (alveolar) | Soft tissue, head/neck, genitourinary | Desmin, MyoD1; t(2;13) alveolar |
Neuroblastoma & Wilms Tumour
Neuroblastoma
Arises from neural crest-derived sympathetic neuroblasts; most common extracranial solid tumour of childhood. 70% arise in the adrenal medulla; rest from paraspinal sympathetic ganglia.
Clinical: abdominal mass crossing midline, hypertension (catecholamine excess → raised VMA/HVA in urine), opsoclonus-myoclonus syndrome (paraneoplastic), periorbital ecchymoses ("raccoon eyes" from retro-orbital metastases).
Morphology: small round blue cells with neuritic processes; Homer-Wright rosettes (tumour cells arranged around a central fibrillary core — not a true lumen).
Molecular: N-myc (MYCN) amplification → poor prognosis; >10 copies = high-risk. Deletion 1p, gain 17q also adverse.
Prognosis paradox: infants (<18 months) have excellent prognosis even with metastases (Stage 4S — spontaneous regression). Older children with N-myc amplification fare poorly.
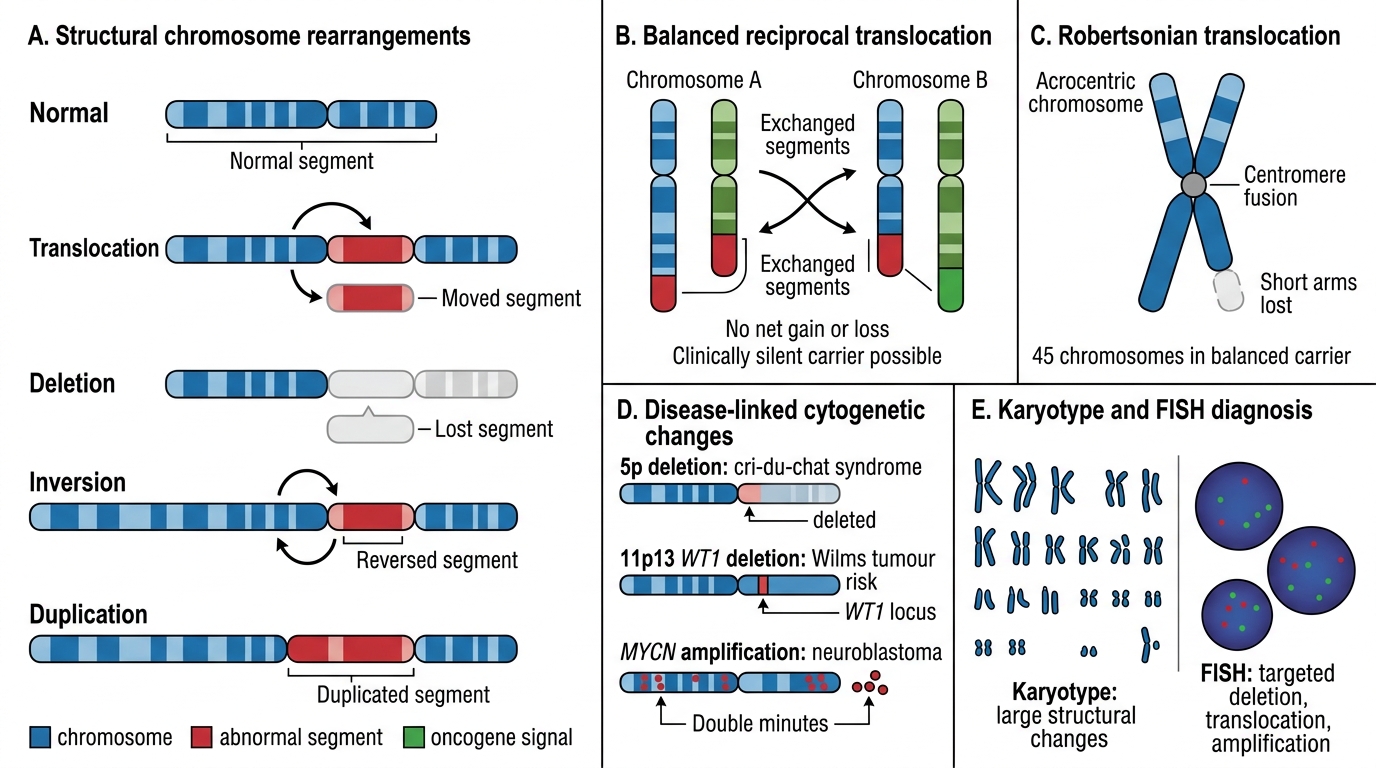
Structural Cytogenetic Abnormalities and Diagnostic Tests
Wilms Tumour (Nephroblastoma)
Arises from metanephric blastema (primitive renal precursor cells). Linked to mutations in WT1 (Wilms tumour suppressor gene, 11p13) and WT2 (11p15).
Associations: WAGR syndrome (Wilms + Aniridia + Genitourinary anomaly + intellectual disability — all from 11p13 deletion); Beckwith-Wiedemann syndrome (gigantism, macroglossia, omphalocele — WT2 region).
Clinical: unilateral abdominal mass (does NOT cross midline — contrast with neuroblastoma), haematuria, hypertension.
Morphology — triphasic histology (pathognomonic): blastemal cells (small round blue cells) + stromal cells (spindle) + epithelial cells (tubules/glomeruli). All three components reflect the attempted — but disorganised — recapitulation of normal nephrogenesis.
Prognosis: excellent overall (90% cure with nephrectomy + chemo ± radiation); anaplastic variant = adverse.
SELF-CHECK
A 3-year-old has a large abdominal mass that does NOT cross the midline. Biopsy shows blastemal cells, stromal cells, and epithelial tubules. Which molecular alteration is most characteristically associated with this tumour?
A. N-myc amplification
B. WT1 gene mutation
C. RB1 gene loss (two-hit)
D. EWSR1-FLI1 fusion t(11;22)
Reveal Answer
Answer: B. WT1 gene mutation
This is Wilms tumour (nephroblastoma). The triphasic histology (blastemal + stromal + epithelial) is pathognomonic. WT1 (11p13) mutation is the characteristic association. N-myc is neuroblastoma; RB1 is retinoblastoma; EWSR1-FLI1 is Ewing sarcoma.