Page 7 of 20
PA11.1-3 | Genetic & Pediatric Diseases — SDL Guide (Part 3)
Retinoblastoma, Ewing Sarcoma & Rhabdomyosarcoma
Retinoblastoma: Genetics, Clinical Features and Histology
Retinoblastoma
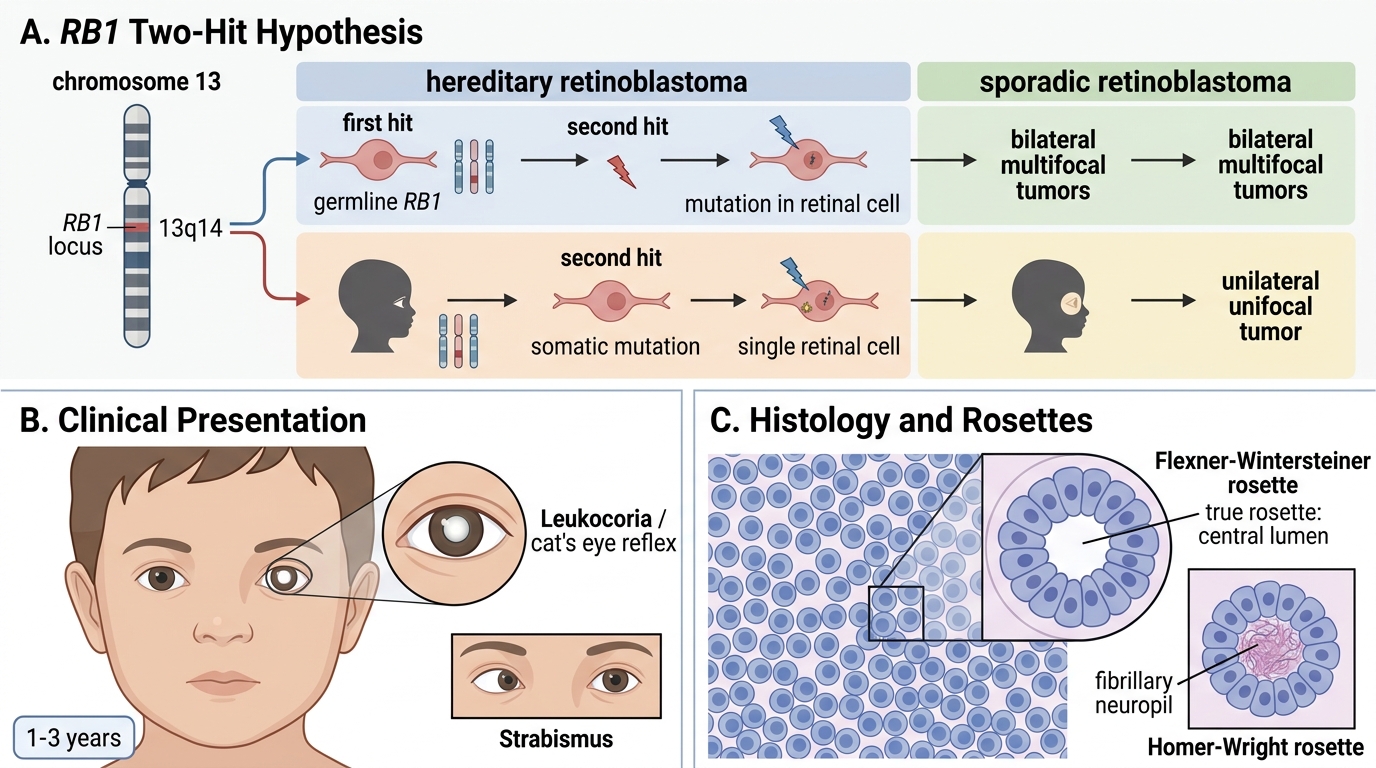
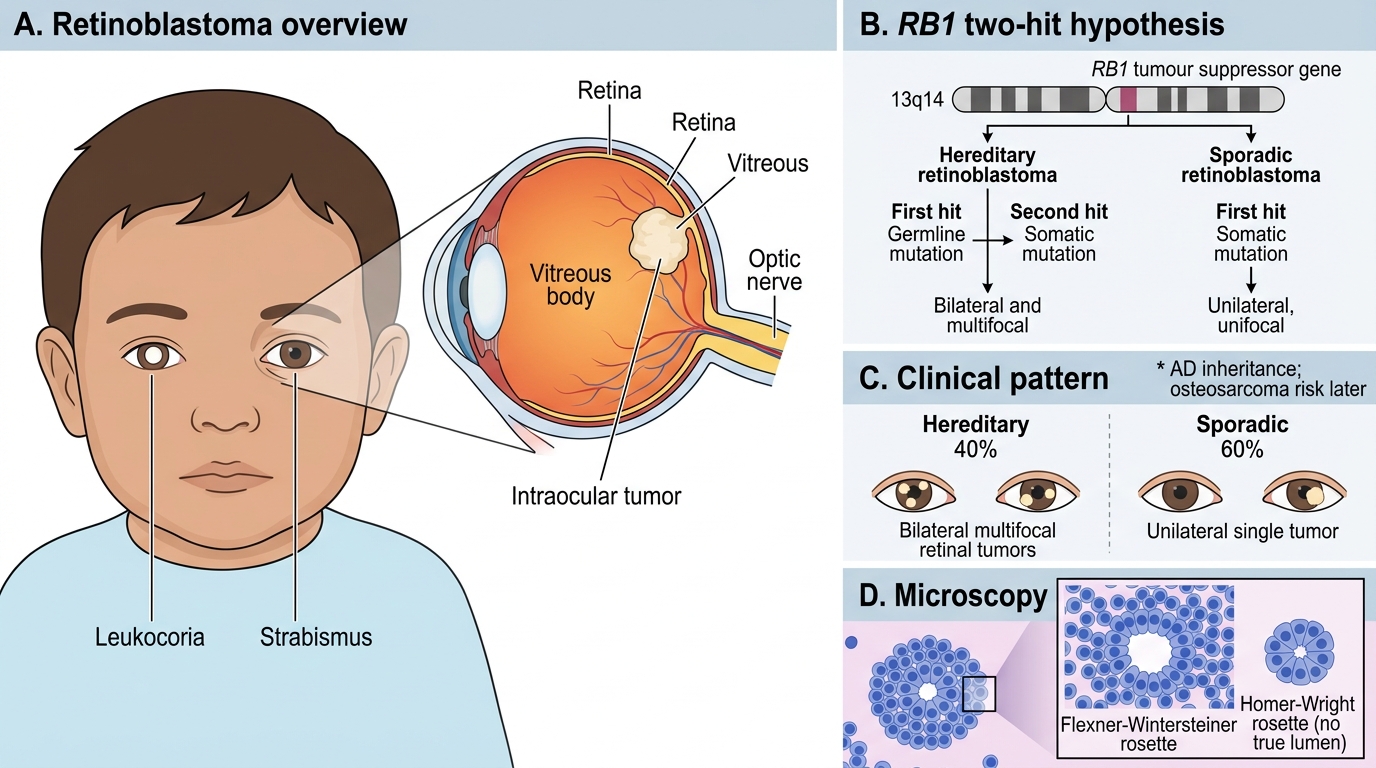
Most common intraocular tumour of childhood. Caused by mutation/deletion of both alleles of the RB1 tumour suppressor gene (chromosome 13q14), the prototype for Knudson's two-hit hypothesis:
- Hereditary (40%): first hit = germline mutation (inherited or de novo), second hit = somatic mutation in any retinal cell → bilateral/multifocal tumours; AD inheritance; also risk of osteosarcoma later.
- Sporadic (60%): both hits somatic → unilateral, unifocal.
Clinical: leukocoria (white pupillary reflex, "cat's eye reflex") in a child aged 1–3 years; strabismus.
Morphology: small round blue cells; Flexner-Wintersteiner rosettes (true rosette — cells surrounding a central lumen, mimicking photoreceptor differentiation); Homer-Wright rosettes also seen.
Ewing Sarcoma
t(11;22)(q24;q12) producing EWSR1-FLI1 fusion gene → aberrant transcription factor. Arises in bone diaphysis (midshaft) of long bones; also chest wall (Askin tumour).
Morphology: monotonous sheets of small round blue cells with clear cytoplasm (glycogen — PAS+); no rosettes. "Onion-skin" periosteal reaction on X-ray.
Rhabdomyosarcoma
Most common soft-tissue sarcoma of childhood; shows skeletal muscle differentiation.
- Embryonal (most common, better prognosis): grape-like (sarcoma botryoides in hollow organs), spindle cells, loss of heterozygosity at 11p15.
- Alveolar (worse prognosis): t(2;13) → PAX3-FOXO1 fusion; alveolar spaces mimicking lung.
Markers: desmin, vimentin, MyoD1 (myogenic transcription factor), myogenin — confirm rhabdomyoblastic differentiation even without cross-striations.
CLINICAL PEARL
"Small round blue cell" differential — your discriminator checklist:
- Site: adrenal/paraspinal → neuroblastoma; kidney → Wilms; retina → retinoblastoma; bone diaphysis → Ewing; soft tissue/head/neck → rhabdomyosarcoma.
- Rosette type: Homer-Wright (neuroblastoma, no lumen) vs. Flexner-Wintersteiner (retinoblastoma, has lumen).
- Urine catecholamines: ↑VMA/HVA → neuroblastoma.
- IHC panel: desmin+/MyoD1+ → rhabdomyosarcoma; NB84/NSE/synaptophysin → neuroblastoma; WT1+ → Wilms blastemal component; CD99 (MIC2)+ → Ewing.
- Key translocation: t(11;22) = Ewing; t(2;13) = alveolar rhabdomyosarcoma.
In a viva, linking histomorphology + molecular marker + translocation for a single tumour earns full marks.
Lysosomal Storage Disorders — Mechanism
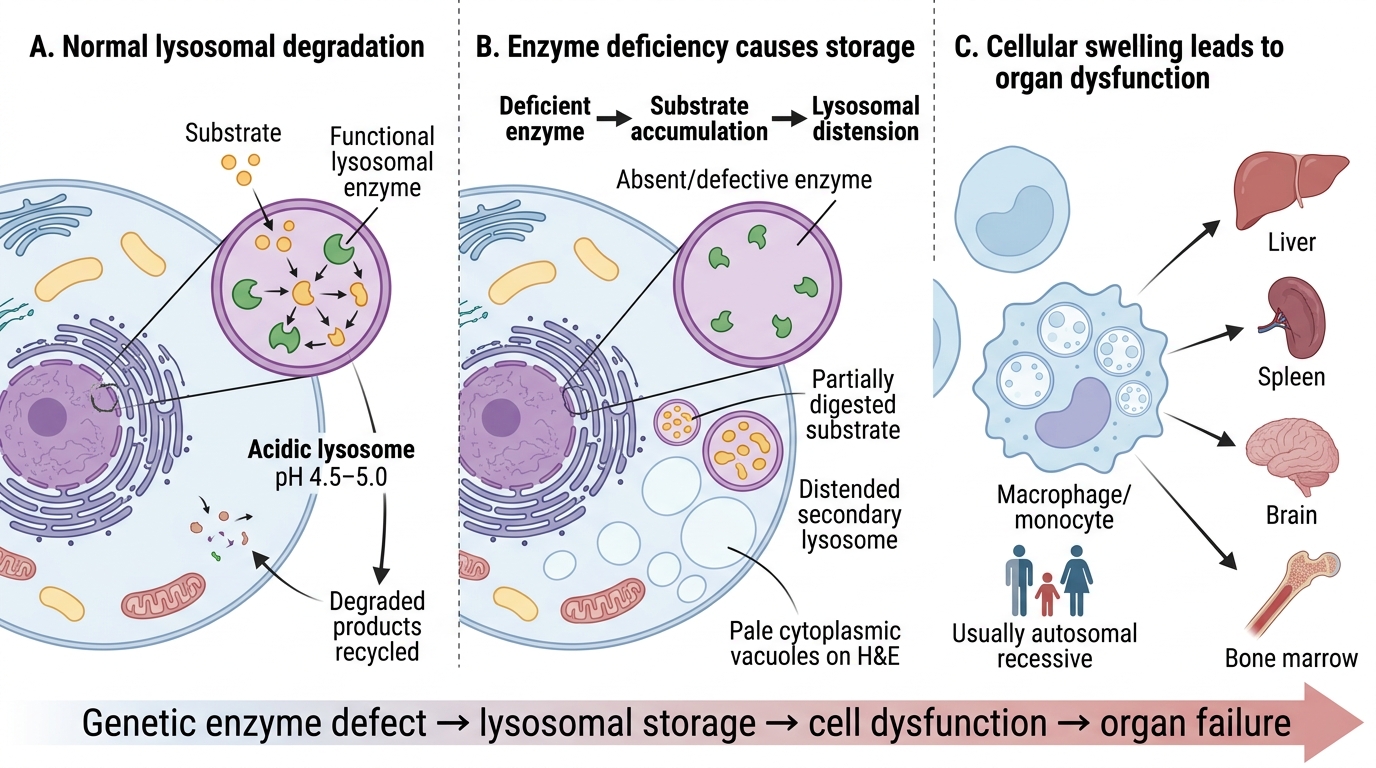
Lysosomal storage disorders (LSDs) share one pathogenic template:
Deficient lysosomal enzyme → substrate accumulates in lysosomes → cell swells and dysfunctions → organ failure
Lysosomes are the cell's recycling plant, using acid hydrolases at pH 4.5–5.0 to degrade glycolipids, glycoproteins, and glycosaminoglycans. When the enzyme is genetically absent or defective:
1. Partially digested substrate (lipid, sugar, protein) packs into secondary lysosomes.
2. Lysosomes distend; the cell's cytoplasm fills with pale vacuoles visible on H&E.
3. Affected cells: primarily macrophages/monocytes (RES) for lipidoses; hepatocytes + neurons for others.
4. Organs most affected: liver, spleen, brain, bone marrow.
Almost all LSDs are autosomal recessive (one functional enzyme copy usually sufficient; disease when both alleles mutant).
Mechanism of Lysosomal Storage Disorders
Gaucher Disease, Niemann-Pick Disease & Tay-Sachs
Gaucher Disease — most common LSD
Deficient enzyme: glucocerebrosidase (acid β-glucosidase) → accumulation of glucocerebroside (glucosylceramide) in macrophages.
Morphology: Gaucher cells — large pale macrophages with a wrinkled "crinkled tissue paper" cytoplasm (glucocerebroside fibrils). Hepatosplenomegaly, bone marrow replacement → pancytopenia, bone pain, pathological fractures, "Erlenmeyer flask" deformity of distal femur on X-ray.
Types: Type 1 (non-neuronopathic, most common, Ashkenazi Jewish population) — no CNS; Types 2 & 3 — CNS involvement.
Treatment: enzyme replacement therapy (imiglucerase) — transforms prognosis of Type 1.
Niemann-Pick Disease
Deficient enzyme: sphingomyelinase → accumulation of sphingomyelin in macrophages and neurons.
Morphology: foam cells (lipid-laden macrophages) in spleen, liver, lymph nodes, bone marrow; neuronal involvement → progressive neurodegeneration. Hepatosplenomegaly + neurodegeneration in Types A/B.
Tay-Sachs Disease
Deficient enzyme: hexosaminidase A (α-subunit mutation) → accumulation of GM2 ganglioside predominantly in neurons.
Clinical: onset 3–6 months; developmental regression, hyperacusis (exaggerated startle to sound), progressive motor/cognitive decline, seizures, death by age 2–4 years.
Pathognomonic finding: cherry-red spot at the macula — the fovea centralis appears red because the surrounding ganglion cell layer (pale from lipid storage) creates a ring that contrasts with the vascular choroid shining through the thin fovea.
Epidemiology: Ashkenazi Jewish population (1:30 carrier rate); AR. No treatment; prenatal diagnosis via amniocentesis.
Retinoblastoma: Genetics, Clinical Features and Microscopy
SELF-CHECK
Bone marrow biopsy from a 30-year-old Ashkenazi Jewish woman with splenomegaly, pancytopenia, and bone pain shows large macrophages with pale wrinkled "crinkled tissue paper" cytoplasm. Which enzyme is deficient?
A. Hexosaminidase A
B. Sphingomyelinase
C. Glucocerebrosidase
D. α-L-iduronidase
Reveal Answer
Answer: C. Glucocerebrosidase
The description is classic for Gaucher cells — large macrophages with wrinkled pale cytoplasm due to glucocerebroside accumulation. The deficient enzyme is glucocerebrosidase (acid β-glucosidase). Hexosaminidase A deficiency = Tay-Sachs (cherry-red spot, neuronal); sphingomyelinase = Niemann-Pick; α-L-iduronidase = Hurler syndrome (MPS I).