Page 8 of 20
PA11.1-3 | Genetic & Pediatric Diseases — SDL Guide (Part 4)
Mucopolysaccharidoses & Glycogen Storage Diseases
Mucopolysaccharidoses and Glycogen Storage Diseases
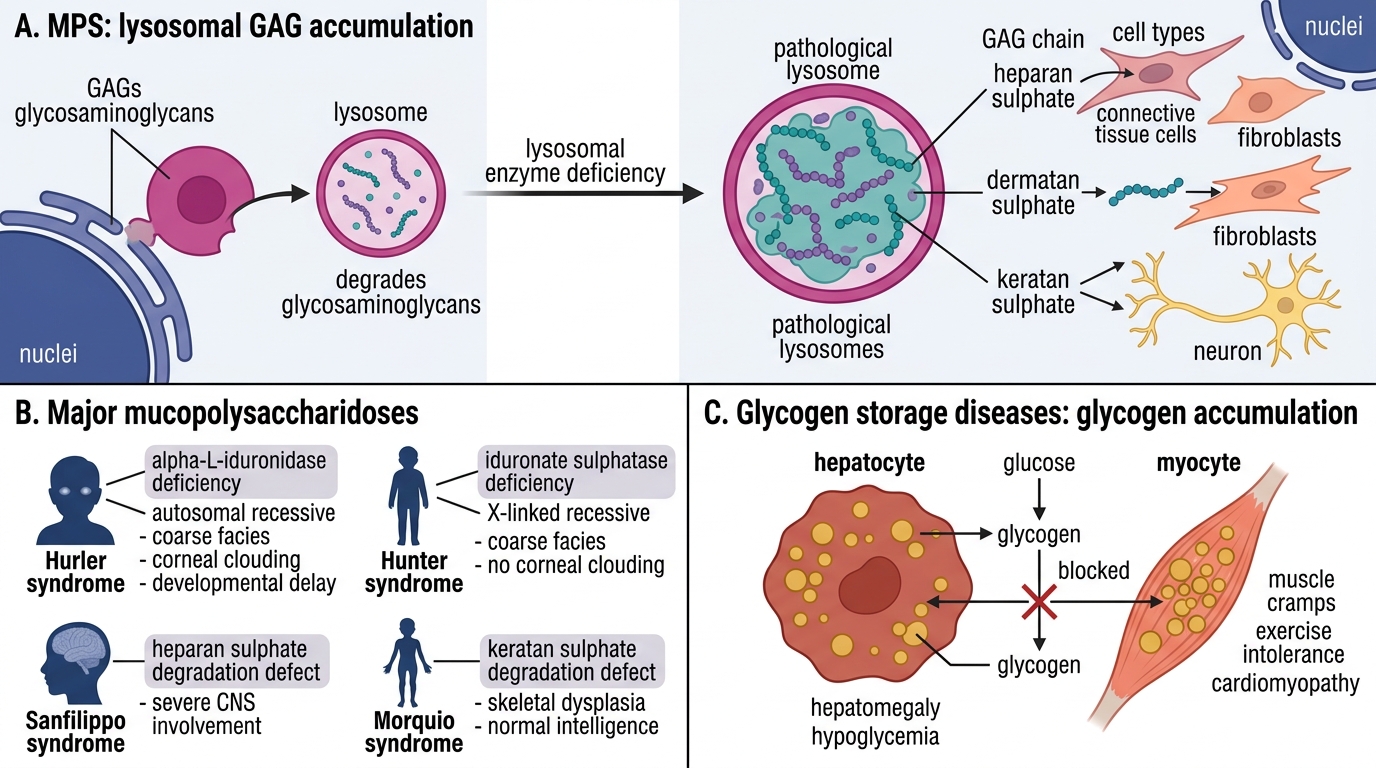
Mucopolysaccharidoses (MPS)
Deficient lysosomal enzymes for degrading glycosaminoglycans (GAGs — heparan, dermatan, keratan sulphates). Undegraded GAGs accumulate in connective tissue cells, fibroblasts, and neurons.
| Disorder | Enzyme deficiency | Inheritance | Key features |
|---|---|---|---|
| Hurler syndrome (MPS I-H) | α-L-iduronidase | AR | Coarse facies (gargoylism), corneal clouding, intellectual disability, hepatosplenomegaly, kyphosis (gibbus), death in childhood |
| Hunter syndrome (MPS II) | Iduronate sulfatase | X-linked recessive | Milder; no corneal clouding; intellectual disability; pebble-skin nodules; survival into adulthood |
Pathology: vacuolated fibroblasts, Kupffer cell swelling; gargoyle cells in spleen. Excess urinary GAGs on screening.
Glycogen Storage Diseases (GSD)
Defects in glycogen synthesis or degradation → glycogen accumulates in specific tissues depending on enzyme:
| Disease | Enzyme | Affected organ | Key feature |
|---|---|---|---|
| von Gierke (GSD type I) | Glucose-6-phosphatase | Liver + kidney | Severe fasting hypoglycaemia, hepatomegaly, doll face, lactic acidosis |
| Pompe disease (GSD type II) | Acid maltase (α-glucosidase) | Heart + skeletal muscle | Massive cardiomegaly, hypotonia, death from cardiorespiratory failure in infancy |
| McArdle (GSD type V) | Myophosphorylase | Skeletal muscle | Exercise-induced muscle cramps; elevated CK; normal lactate with exercise |
SELF-CHECK
An 8-month-old infant has massive cardiomegaly, generalised hypotonia, and dies from cardiorespiratory failure. Biopsy of myocardial cells shows PAS-positive vacuoles. Which enzyme deficiency is responsible?
A. Glucose-6-phosphatase
B. Acid maltase (lysosomal α-glucosidase)
C. Glucocerebrosidase
D. Debranching enzyme
Reveal Answer
Answer: B. Acid maltase (lysosomal α-glucosidase)
Pompe disease (GSD type II) is caused by acid maltase (lysosomal α-glucosidase) deficiency. Glycogen accumulates in lysosomes of cardiac and skeletal muscle → massive cardiomegaly + hypotonia. Glucose-6-phosphatase deficiency = von Gierke (liver/kidney, hypoglycaemia); glucocerebrosidase = Gaucher disease.
CLINICAL PEARL
High-yield LSD memory aid — "GNT MHP":
- Gaucher → Glucocerebrosidase → Gaucher cells (wrinkled macrophages)
- Niemann-Pick → NsphingomyeliNase → foam cells + neurodegeneration
- Tay-Sachs → Texosaminidase A → cherry-red spot (neurons)
- Mucopolysaccharidoses → Multiple GAG-enzyme defects → MPS I (Hurler, AR) / MPS II (Hunter, X-linked)
- Hex from Hurler = Heparan + dermatan; Hunter = no corneal clouding
- Pompe → Pump failure (heart); von Gierke → Glucose-6-P = Gluconeogenesis failure → hypoglycaemia
Note: Pompe is unique — the only GSD that is also a lysosomal storage disorder (acid maltase is a lysosomal enzyme).