Page 1 of 20
PA10.1-5 | Infections & Infestations — SDL Guide
Learning Objectives
- Describe the general principles of host–pathogen interaction, routes of infection, and the spectrum of tissue responses to infection.
- Outline the life cycle of Plasmodium falciparum, the pathogenesis of cerebral malaria, and the morphological features seen on peripheral blood smear.
- Explain the clinico-pathological basis of the tuberculoid–lepromatous spectrum in leprosy, correlating immune response with lesion type.
- Describe the pathology of neurocysticercosis caused by Taenia solium.
- Identify the characteristic tissue reaction patterns of common bacterial, viral, protozoal, fungal, and helminthic infections.
- Explain the pathogenesis of COVID-19, including diffuse alveolar damage, cytokine storm, and coagulopathy, alongside key laboratory findings.
INSTRUCTIONS
Infectious disease kills more people globally than any other cause. As a pathologist, you are the one who confirms whether a brain lesion is a parasite, a granuloma, or a tumour — and that confirmation changes the entire management. This module builds a systematic framework for reading tissue responses: once you know that a granuloma means 'the macrophage is trapped', that foamy lepra cells mean 'the bacterium is winning', and that diffuse alveolar damage means 'the lung barrier has failed', you can apply the same logic to any new pathogen you encounter in your career. Work through each section in sequence; the micro-quizzes test whether you can distinguish look-alike lesions under examination pressure.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch 8 (Infectious Diseases) (textbook)
- Harsh Mohan Textbook of Pathology, 8th ed., Ch 5 (Infectious and Parasitic Diseases) (textbook)
- WHO Global Tuberculosis Report 2023 (leprosy epidemiology section) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 28-year-old tribal health worker returns from Chhattisgarh with a 5-day history of cyclic fever every 48 hours, severe headache, and confusion. His blood film shows infected RBCs with multiple ring forms and 'appliqué' (accolé) positions. A 35-year-old from rural Bihar has new-onset seizures; MRI shows ring-enhancing lesions in the brain. A leprosy patient in Tamil Nadu has lost sensation in his hands and has thickened nerves. Three patients — three parasites — three completely different tissue stories. By the end of this module, you will be able to explain exactly what the pathogen is doing to the host tissue in each case, and why.
WHY THIS MATTERS
India carries one of the highest burdens of infectious disease in the world: malaria kills thousands annually (falciparum predominating in tribal belts), India eliminated leprosy as a public health problem in 2005 but still reports 100,000+ new cases per year, and cysticercosis is the leading preventable cause of adult-onset epilepsy in the country. COVID-19 reshaped how we understand lung pathology and coagulopathy. The NMC CBUC 2024 expects you to correlate clinical features with pathological mechanisms — not merely list causative organisms. These are examination favourites and life-saving clinical skills.
RECALL
Before you start, activate what you already know:
- From Year-1 Microbiology: Plasmodium species and their mosquito vectors; Mycobacterium leprae staining (AFB); Taenia solium life cycle.
- From Year-1 Physiology: How the spleen filters abnormal RBCs; the role of macrophages in phagocytosis.
- From Year-1 Biochemistry: What haemoglobin breakdown products are; why unconjugated bilirubin rises in haemolysis.
- From Pathology Block 1: The difference between granulomatous and suppurative inflammation; what a Langerhans giant cell is.
If any of these feel fuzzy, do a 3-minute mental recall before continuing — it will make the mechanistic explanations below much sharper.
General Principles: Host–Pathogen Interaction
Every infectious disease is a negotiation between the pathogen's virulence and the host's defence. Pathogenicity is the ability of an organism to cause disease; virulence is the degree of that ability, determined by factors such as toxin production, adhesion molecules, capsule (anti-phagocytic), and intracellular survival.
Routes of entry define which tissues are at risk:
• Respiratory (airborne/droplet) — lungs → tuberculosis, COVID-19, measles
• Faeco-oral — gut → typhoid, cholera, polio
• Skin/vector-borne — blood → malaria, dengue, Lyme disease
• Sexual/vertical — systemic → HIV, syphilis, congenital rubella
Once inside, the tissue response depends on the pathogen class:
| Pathogen type | Predominant tissue reaction |
|---|---|
| Pyogenic bacteria | Suppurative (neutrophilic) inflammation; abscess |
| Mycobacteria, fungi | Granulomatous inflammation |
| Viruses | Cytopathic effects; intranuclear/cytoplasmic inclusions; lymphocytic infiltrate |
| Parasites (helminths) | Eosinophilic inflammation; Charcot–Leyden crystals |
| Protozoa | Variable — haemolysis (malaria), necrosis (amoeba), cyst formation |
Host factors that amplify disease: immunosuppression, malnutrition, co-infection, and genetic polymorphisms (e.g., HbS heterozygotes are partially protected against falciparum malaria — a classical board question).
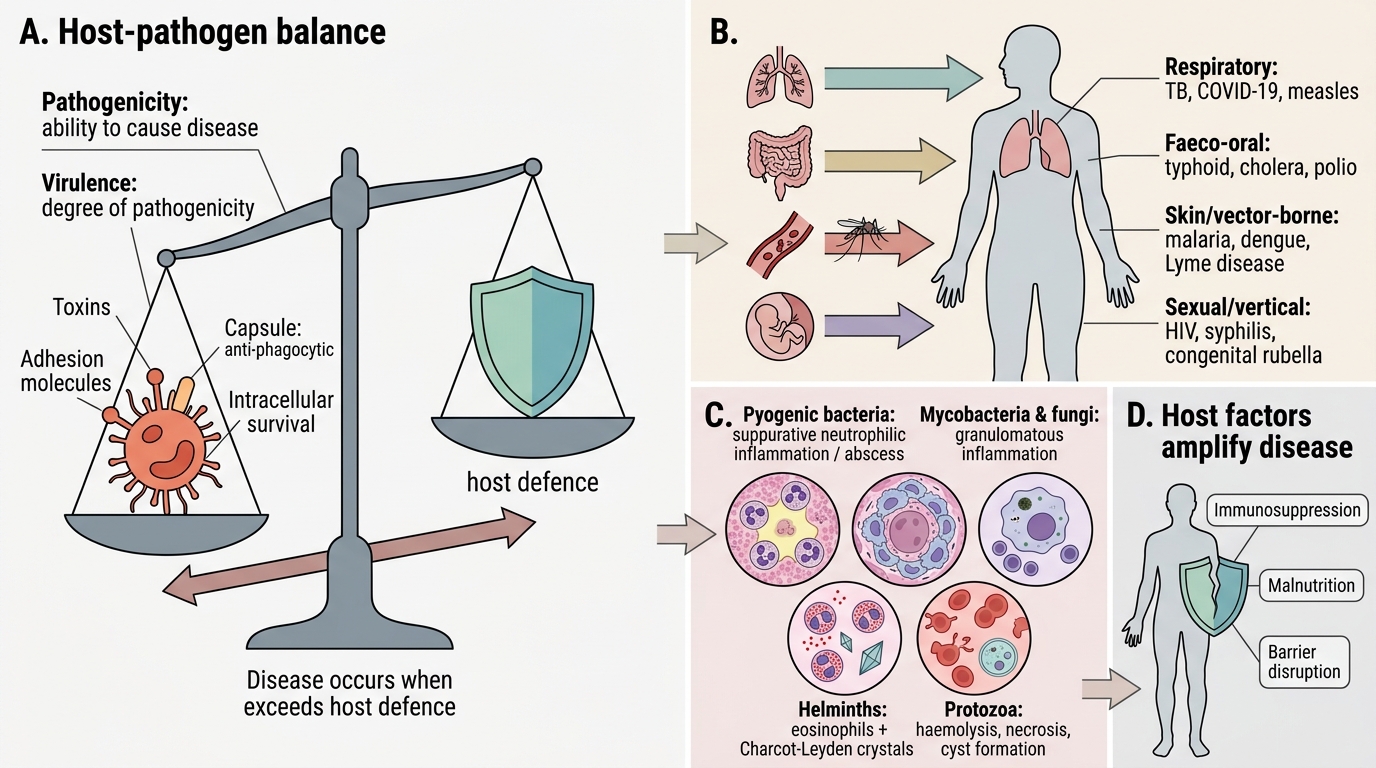
Host-Pathogen Interaction: Entry, Virulence, Tissue Response
Malaria: Life Cycle, Pathogenesis, and Pathology
Malaria is caused by Plasmodium species transmitted by the female Anopheles mosquito. Four species infect humans: P. falciparum (most lethal), P. vivax, P. ovale, P. malariae. P. falciparum causes almost all severe/cerebral malaria.
Life cycle (key steps for pathology):
1. Mosquito injects sporozoites → travel to liver → liver-stage schizogony (hepatocytes; clinically silent; P. vivax/ovale form dormant hypnozoites)
2. Merozoites released → invade RBCs → erythrocytic schizogony
3. Ring form → trophozoite → schizont → rupture (releasing merozoites + pyrogenic toxins every 48 h for falciparum/vivax/ovale, 72 h for malariae) → cyclic fever
4. Some merozoites → gametocytes (ingested by mosquito → sexual cycle)
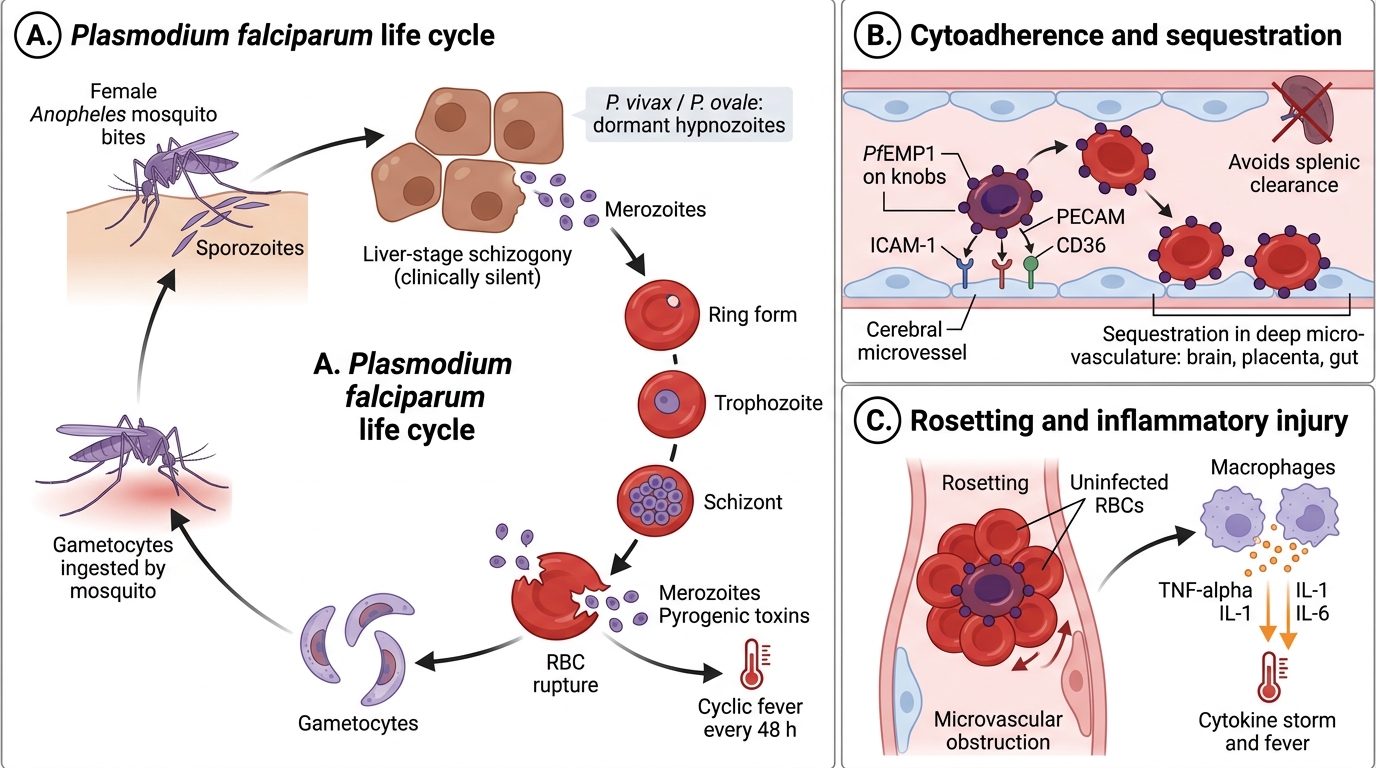
Falciparum Malaria: Life Cycle and Severe Disease Mechanisms
Pathogenesis of severe falciparum malaria:
• Cytoadherence/sequestration: P. falciparum-infected RBCs express PfEMP1 (P. falciparum erythrocyte membrane protein 1) on knobs → bind endothelial ICAM-1, PECAM, CD36 → infected RBCs sequester in deep microvasculature (brain, placenta, gut) — they never circulate to the spleen for destruction
• Rosetting: infected RBCs bind uninfected RBCs → microvascular obstruction
• Cytokine storm: TNF-α, IL-1, IL-6 released by macrophages → fever, hypoglycaemia, metabolic acidosis
• Cerebral malaria: sequestration in cerebral capillaries → ring haemorrhages (Dürck's granulomas — microglial nodules around necrotic vessels), coma; mortality 15–20% even with treatment
Pathological features:
• Spleen: massive splenomegaly; pulp congestion; haemosiderin-laden macrophages; malarial pigment (haemozoin) — black-brown granular pigment from HbF degradation, seen in macrophages
• Liver: hepatomegaly; Kupffer cells packed with haemozoin; centrizonal necrosis in severe cases
• Kidneys: in P. malariae — immune complex GN → nephrotic syndrome; in falciparum — blackwater fever (massive intravascular haemolysis → haemoglobinuria → acute tubular necrosis → AKI)
• Brain: cerebral malaria; ring haemorrhages; cerebral oedema
Smear diagnosis (must know):
| Feature | P. falciparum | P. vivax |
|---|---|---|
| Ring forms | Multiple per RBC; 'appliqué'/'accolé' | Usually one per RBC |
| Schizont in peripheral smear | Absent (sequestered) | Present |
| Infected RBC size | Normal | Enlarged |
| Gametocyte | Crescent/banana-shaped | Round |
| Schüffner's dots | Absent | Present |
SELF-CHECK
A peripheral blood smear shows RBCs of normal size, multiple ring forms per cell including 'appliqué' forms, and crescent-shaped gametocytes. No Schüffner's dots are seen and no schizonts are visible. Which Plasmodium species is MOST likely, and why are schizonts absent from the peripheral blood?
A. P. vivax — schizonts are selectively destroyed by the complement system
B. P. malariae — 72-hour cycle means schizonts are too rare to detect
C. P. falciparum — infected RBCs sequester in deep microvasculature via PfEMP1-mediated cytoadherence
D. P. ovale — Schüffner's dots are absent in early infections
Reveal Answer
Answer: C. P. falciparum — infected RBCs sequester in deep microvasculature via PfEMP1-mediated cytoadherence
The crescent gametocytes, multiple rings per RBC, normal RBC size, and absence of Schüffner's dots are all hallmarks of P. falciparum. Schizonts are absent from peripheral blood because P. falciparum-infected RBCs express PfEMP1 on surface knobs, which bind endothelial receptors (ICAM-1, CD36) in deep capillaries — this sequestration prevents mature forms from circulating to the spleen or appearing in a peripheral smear. P. vivax schizonts DO appear in peripheral blood, and P. vivax/ovale both show Schüffner's dots.
Cysticercosis: Taenia solium and Neurocysticercosis
Cysticercosis is caused by the larval (cysticercus) stage of Taenia solium (pork tapeworm). It is the most common helminthic infection of the nervous system and the leading cause of acquired epilepsy in developing countries.
How infection occurs:
• Humans are the definitive host (adult tapeworm in intestine — taeniasis) and may also be accidental intermediate hosts (cysticercosis)
• Ingestion of T. solium eggs (from contaminated food/water, or autoinfection via faeco-oral route) → oncospheres hatch → penetrate gut wall → disseminate haematogenously → encyst in tissues
Sites of cyst deposition: Brain (most important) > muscle > eye > subcutaneous tissue > liver
Pathology of the cysticercus:
• The larva develops into a fluid-filled cysticercus — a translucent cyst (0.5–2 cm) containing a scolex (invaginated head with suckers and hooklets) → visible on MRI as a 'cyst with an eccentric scolex' (pathognomonic)
• Initially, the cyst wall elicits little host reaction (the live larva is immunologically tolerated)
• When the larva dies → intense eosinophilic/granulomatous host reaction → surrounding oedema → seizures, raised ICP
• Over time: cyst calcifies → calcified nodule on CT (classic finding in old neurocysticercosis)
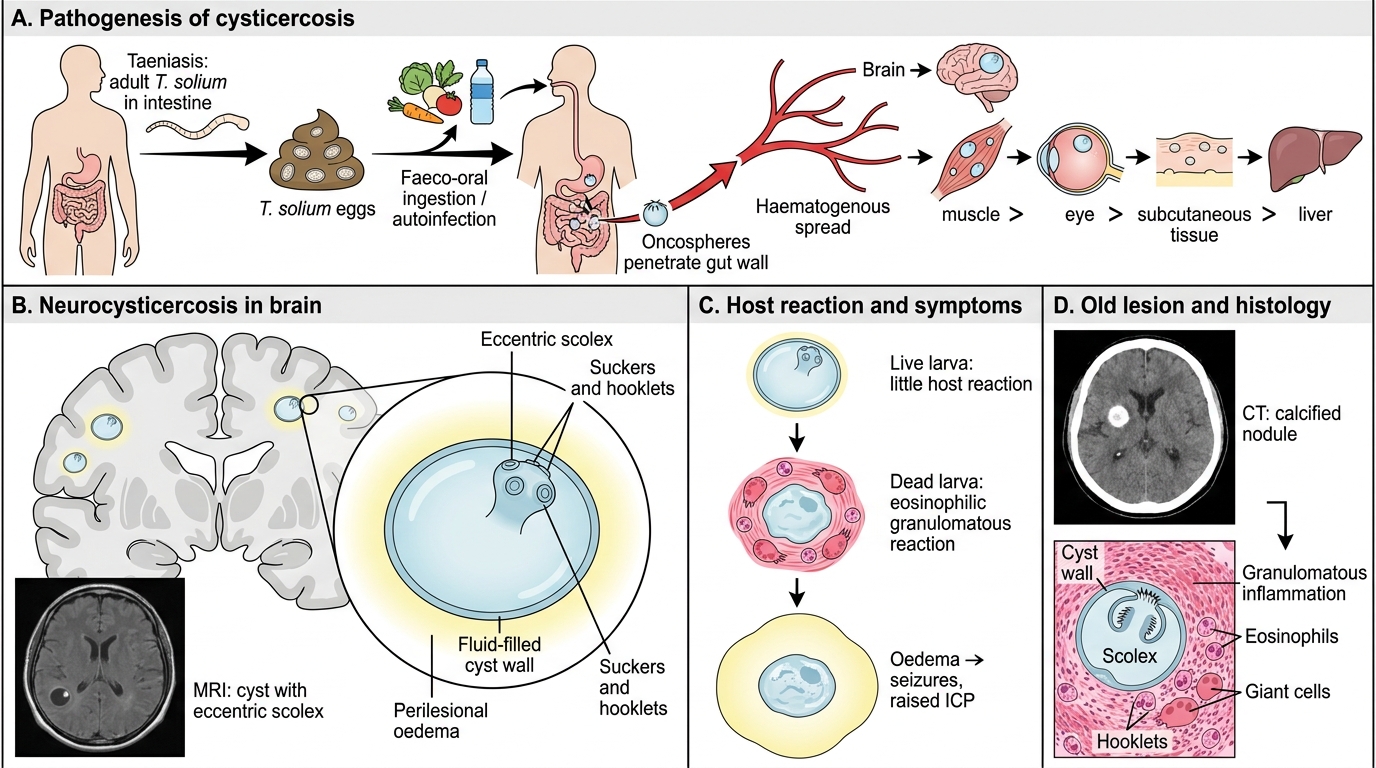
Cysticercosis and Neurocysticercosis
Stages of neurocysticercosis on imaging (correlates with pathology):
1. Vesicular stage — live cyst, no oedema, minimal inflammation
2. Colloidal stage — cyst fluid becomes turbid, significant oedema (symptomatic)
3. Granular/nodular stage — cyst shrinks, granulomatous inflammation
4. Calcified stage — dead, calcified nodule
Clinical features: new-onset seizures (most common), focal neurological deficits, raised ICP (hydrocephalus), meningitis (subarachnoid NCC)
Diagnosis: MRI (cyst with scolex), CT (calcifications), serology (ELISA for cysticercal antibodies), CSF — eosinophilic pleocytosis
Clinical pearl: A young adult from a rural background presenting with new seizures and a ring-enhancing brain lesion on CT/MRI — always consider neurocysticercosis before tuberculoma or glioma.