Page 2 of 20
PA10.1-5 | Infections & Infestations — SDL Guide (Part 2)
Leprosy: The Tuberculoid–Lepromatous Spectrum
Leprosy (Hansen's disease) is caused by Mycobacterium leprae, an obligate intracellular organism that infects peripheral nerves and skin macrophages. It is transmitted via prolonged close contact (nasal droplets). India accounts for ~50% of global cases.
The key concept: the Ridley–Jopling spectrum
The clinical and pathological presentation is entirely determined by the host's cell-mediated immune (CMI) response — not the bacterium's intrinsic virulence.
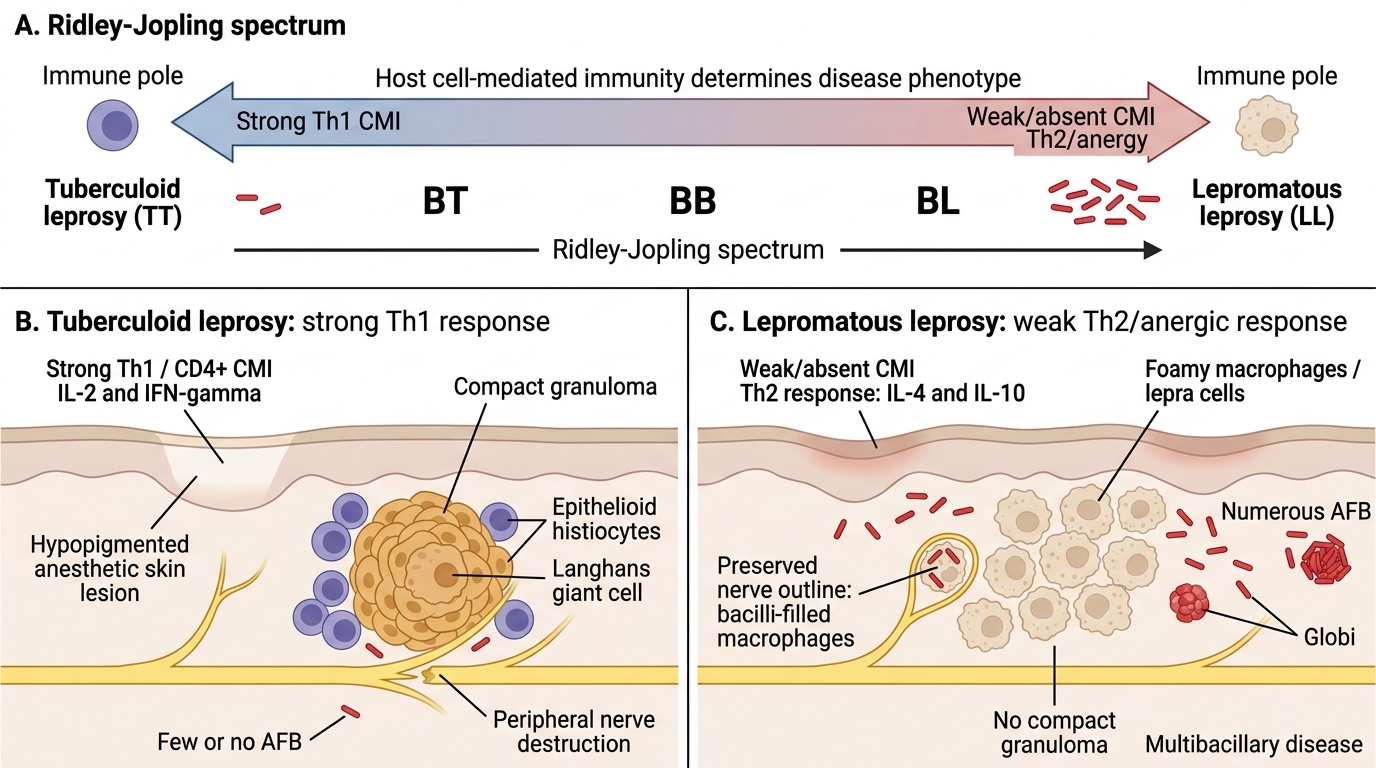
Leprosy: Tuberculoid-Lepromatous Spectrum
| Feature | Tuberculoid (TT) | Lepromatous (LL) |
|---|---|---|
| CMI response | Strong (Th1, CD4+, IL-2, IFN-γ) | Weak/absent (Th2, IL-4, IL-10 anergy) |
| Bacterial load | Paucibacillary — few/no AFB | Multibacillary — numerous AFB |
| Skin lesions | Few (1–5), well-defined, hypopigmented, anaesthetic | Many, diffuse, poorly-defined, nodular; leonine facies |
| Nerve involvement | Asymmetric, marked; single enlarged nerve; early loss of sensation | Symmetric, glove-and-stocking; gradual |
| Histology | Compact epithelioid granulomas with Langerhans giant cells, lymphocytic cuff; few/no AFB on Fite stain | Foamy macrophages (lepra cells/Virchow cells) packed with AFB (globi); no true granuloma; lymphocytes scarce |
| Lepromin test | Strongly positive (Fernandez + Mitsuda) | Negative |
| Infectivity | Low | High |
Borderline leprosy (BT, BB, BL): intermediate forms; immunologically unstable; most common clinically.
Nerve pathology (critical for exam):
• M. leprae has unique tropism for Schwann cells (peripheral nerve) and cooler tissues (skin, testes, anterior eye)
• In TT: granulomatous destruction of nerve → early, severe, focal neuropathy → claw hand, foot drop, wrist drop
• In LL: infiltration of nerve by bacillus-laden macrophages → gradual demyelination → glove-and-stocking sensory loss → painless injuries → trophic ulcers, Charcot joint
• Nerves to know: ulnar (claw hand), median (ape hand), radial (wrist drop), common peroneal (foot drop), great auricular, posterior tibial, facial nerve
Diagnosis:
• Slit-skin smear: scraping from earlobe, elbow skin, lesion edge → Fite stain (modified acid-fast) → Bacterial Index (BI): 0 (TT) to 6+ (LL)
• Skin biopsy: definitive — shows granuloma vs. foamy macrophages
• Lepromin test: measures CMI, NOT a diagnostic test for active disease
Lepra reactions (acute inflammatory episodes):
• Type 1 (reversal reaction): CMI suddenly upregulates in borderline leprosy → oedematous skin lesions + acute nerve swelling → risk of permanent nerve damage
• Type 2 (Erythema Nodosum Leprosum, ENL): immune-complex-mediated (Type III hypersensitivity) in multibacillary disease → painful red nodules, fever, neuritis, iritis
SELF-CHECK
A biopsy from a patient with leprosy shows the dermal nerves are damaged and surrounded by compact epithelioid granulomas with Langerhans giant cells and a dense lymphocytic cuff. Fite stain shows NO acid-fast bacilli. The lepromin test is strongly positive. Which type of leprosy does this represent, and what is the dominant cytokine driving this immune response?
A. Lepromatous leprosy (LL); IL-4 and IL-10 from Th2 cells
B. Tuberculoid leprosy (TT); IFN-γ and IL-2 from Th1 cells
C. Borderline lepromatous (BL); TGF-β from regulatory T cells
D. Indeterminate leprosy; IFN-γ from NK cells only
Reveal Answer
Answer: B. Tuberculoid leprosy (TT); IFN-γ and IL-2 from Th1 cells
Compact epithelioid granulomas, Langerhans giant cells, lymphocytic cuff, absent AFB, and a positive lepromin test are the hallmarks of tuberculoid leprosy (TT). The strong CMI response is Th1-driven: CD4+ T cells produce IFN-γ (activates macrophages to form granulomas and kill bacilli) and IL-2 (promotes T-cell proliferation). This is why bacterial load is low and the tissue response is vigorous — the immune system is winning. In LL, Th2 dominance (IL-4/IL-10) suppresses CMI → anergy → bacilli multiply unchecked inside foamy macrophages.
CLINICAL PEARL
The 'paradox of leprosy' in a single sentence: The patient who mounts the best immune response (TT) suffers the worst nerve damage — because the granuloma itself destroys the nerve in its attempt to contain the bacillus. The patient who mounts no immune response (LL) preserves nerve architecture longer but becomes a reservoir of infection with millions of bacilli in the skin. This paradox illustrates a universal principle in pathology: the host's inflammatory response is simultaneously protective and destructive.
Common Infections — Tissue Reaction Patterns
Understanding the dominant tissue reaction for each pathogen class allows you to narrow a differential diagnosis from histology alone — a core clinical pathology skill.
Suppurative (neutrophilic) infections:
• Caused by pyogenic bacteria: Staphylococcus aureus, Streptococcus pyogenes, Klebsiella, Pseudomonas
• Tissue reaction: abscess (central liquefactive necrosis + neutrophil pus + fibrous wall) or empyema (pus in body cavity)
• Examples: lung abscess, cerebral abscess, osteomyelitis (Brodie's abscess)
Granulomatous infections:
• Caused by mycobacteria (TB, leprosy), fungi (Histoplasma, Cryptococcus), parasites (schistosome eggs)
• Tissue reaction: epithelioid granuloma ± central caseous necrosis (TB) or non-caseating (sarcoid, fungi)
• TB-specific features: caseation + Langhans giant cells (peripheral nuclear arrangement) + Ghon focus (lung primary) + Ranke complex (calcified Ghon + hilar node)
Cytopathic/viral infections:
• Viruses cause cytopathic effects visible on histology:
| Virus | Inclusion body | Location |
|---|---|---|
| CMV | 'Owl-eye' intranuclear + cytoplasmic | Endothelial cells, pneumocytes |
| HSV | Cowdry type A intranuclear | Neurons, hepatocytes |
| Rabies | Negri bodies (eosinophilic cytoplasmic) | Hippocampal neurons |
| Measles | Warthin–Finkeldey giant cells | Lymph nodes |
| Molluscum | Henderson–Paterson bodies | Keratinocytes |
Helminthic infections (eosinophilic response):
• Tissue eosinophilia is the hallmark of helminth (worm) infections
• Charcot–Leyden crystals: breakdown products of eosinophil granules; hexagonal, elongated crystals found in stool, sputum, tissue
• Examples: Ascaris (granulomatous in liver), Schistosoma (hepatic fibrosis — Symmers' pipe-stem fibrosis, granulomas around eggs), Strongyloides (larva in gut mucosa)
Fungal infections:
• Dimorphic fungi (Histoplasma, Blastomyces, Coccidioides): granulomatous reaction — especially in immunocompetent
• Opportunistic fungi (Candida, Aspergillus, Mucor): suppurative + necrotizing + vascular invasion in immunosuppressed
• Cryptococcus neoformans: meningoencephalitis; mucoid capsule → 'soap-bubble' lesions on MRI; India ink preparation (lab); PAS/mucicarmine stain (histology)
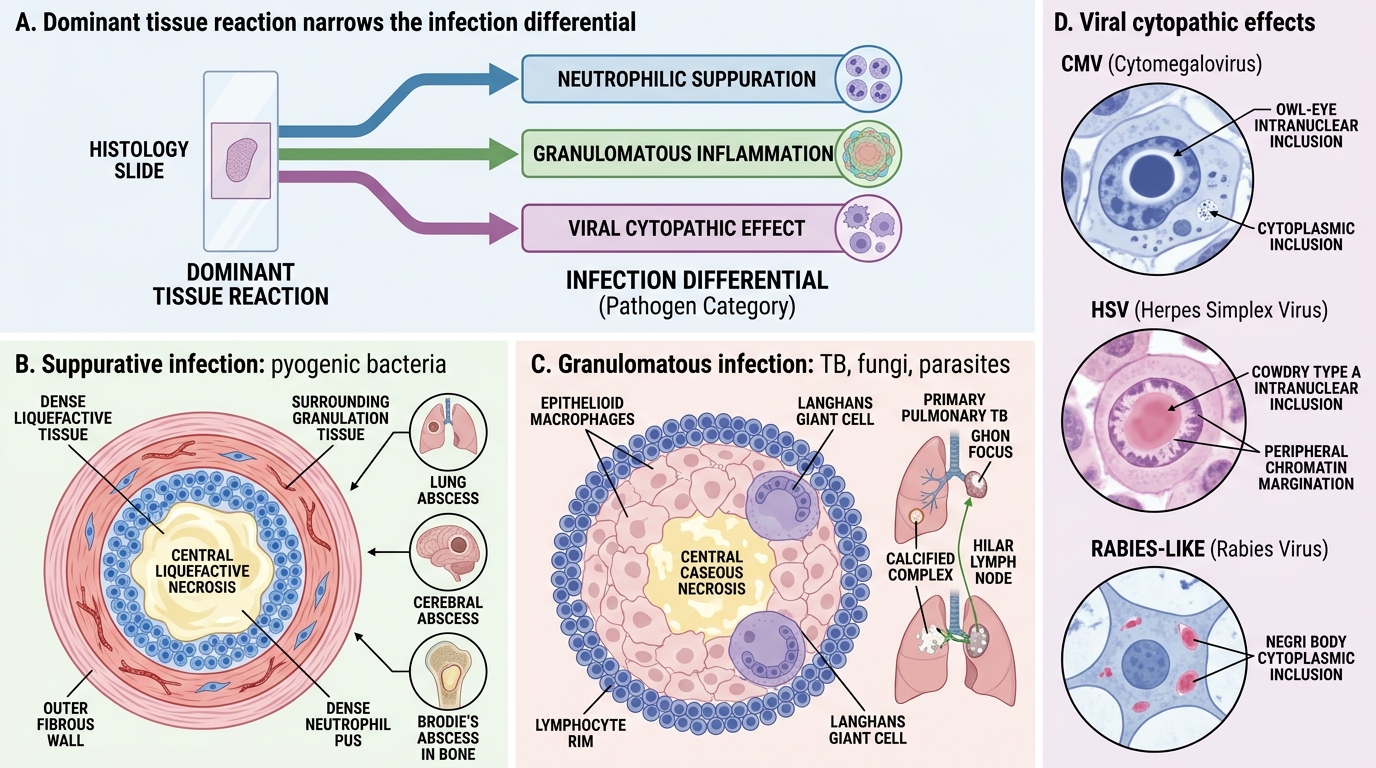
Common Infections: Tissue Reaction Patterns
SELF-CHECK
A liver biopsy from a patient with chronic schistosomiasis shows extensive periportal fibrosis with pale, thickened portal tracts ('pipe-stem' pattern) and granulomas containing eosinophilic debris. Portal hypertension has developed. Which term describes this characteristic hepatic fibrosis pattern, and what is its mechanism?
A. Bridging fibrosis; due to direct hepatocyte necrosis by schistosomal toxins
B. Symmers' pipe-stem fibrosis; due to granulomatous reaction to schistosome eggs lodged in portal venules, leading to progressive periportal fibrosis
C. Nutmeg liver; due to chronic passive venous congestion in portal hypertension
D. Mallory–Denk body formation; due to alcoholic hepatitis co-existing with schistosomiasis
Reveal Answer
Answer: B. Symmers' pipe-stem fibrosis; due to granulomatous reaction to schistosome eggs lodged in portal venules, leading to progressive periportal fibrosis
Symmers' pipe-stem (clay-pipe-stem) fibrosis is the pathological hallmark of hepatosplenic schistosomiasis (S. mansoni/japonicum). Schistosome eggs released into the portal circulation lodge in portal venules → trigger a vigorous granulomatous reaction (eosinophilic granuloma around the egg) → progressive periportal fibrosis → portal hypertension. Critically, hepatocyte architecture is largely preserved (unlike cirrhosis), so liver synthetic function is relatively maintained for longer — but variceal bleeding is the major complication. The 'pipe-stem' refers to the pale, thickened portal tracts resembling a clay pipe stem on gross pathology.
COVID-19: Pathogenesis, Lung Pathology, and Laboratory Findings
COVID-19 is caused by SARS-CoV-2, a betacoronavirus. It emerged in Wuhan, China (December 2019) and caused a global pandemic. Understanding its pathology explains the clinical features and lab abnormalities seen in severe disease.
Entry mechanism:
• SARS-CoV-2 uses its spike (S) protein to bind ACE2 (angiotensin-converting enzyme 2) on host cells
• ACE2 is expressed on type II pneumocytes (lung), enterocytes (gut), renal tubular cells, endothelium, and cardiomyocytes — explaining the multi-organ involvement
• TMPRSS2 (transmembrane serine protease 2) primes the spike protein for fusion → viral entry
Pathogenesis of severe COVID-19 (three overlapping mechanisms):
1. Diffuse Alveolar Damage (DAD): the cardinal lung lesion
• Direct viral cytopathic effect on type II pneumocytes → denudation of alveolar epithelium
• Exudative phase (days 1–7): alveolar oedema, fibrin, protein-rich fluid → hyaline membrane formation (eosinophilic membranes lining alveolar walls) → clinically = ARDS
• Proliferative phase (days 7–21): type II pneumocyte hyperplasia (reactive, cuboidal cells lining alveoli), fibroblast proliferation
• Organising phase: interstitial fibrosis (may be permanent)
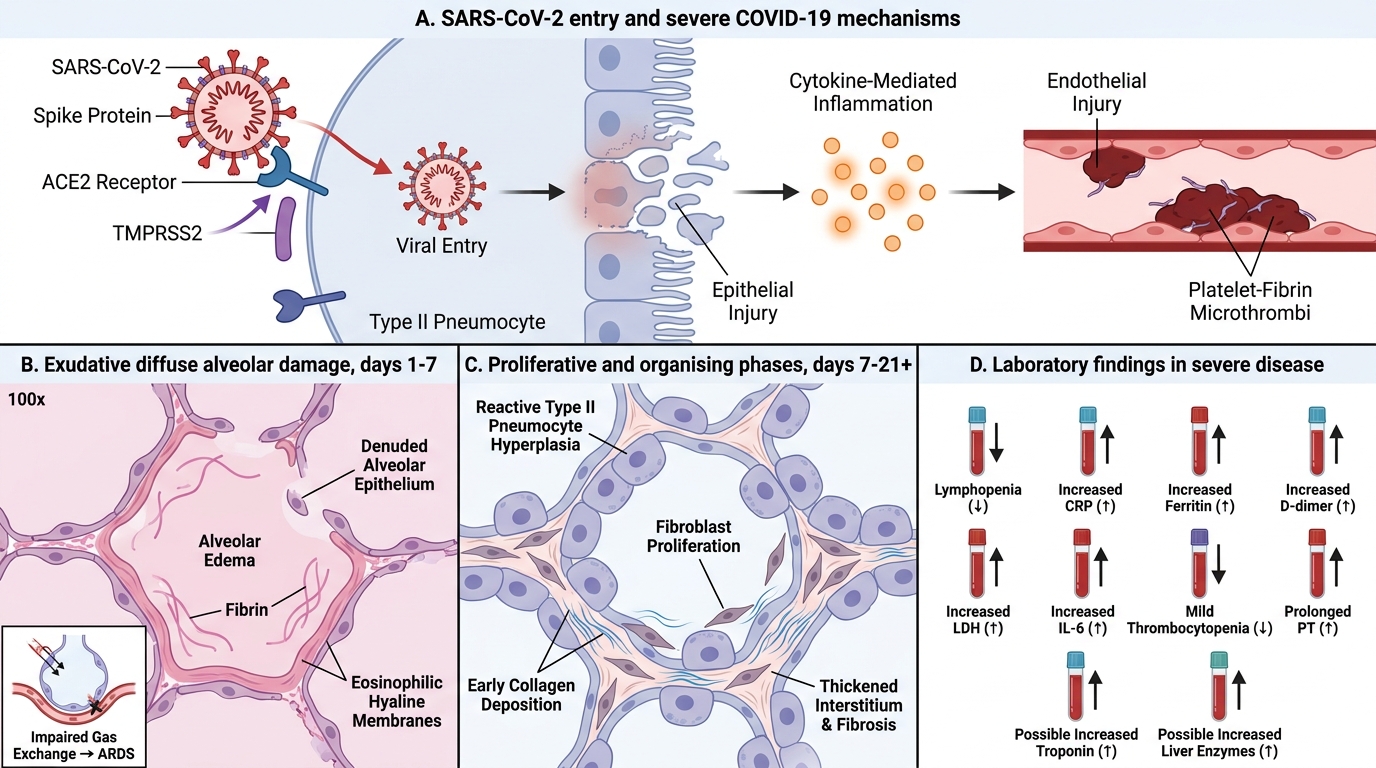
COVID-19 Pathogenesis, Lung Pathology, and Laboratory Findings
2. Cytokine storm (hyperinflammation):
• Dysregulated innate immune response → massive release of pro-inflammatory cytokines: IL-6, TNF-α, IL-1β, IL-8
• Amplified by impaired type I interferon response (SARS-CoV-2 actively suppresses IFN-α/β)
• Result: systemic endothelial activation, capillary leak, multi-organ dysfunction
• IL-6 is the key cytokine — elevated markedly in severe disease; IL-6 receptor antagonists (tocilizumab) are now standard therapy
3. Coagulopathy and microthrombi:
• Endothelial injury (direct viral infection + cytokine-mediated) → exposure of subendothelial collagen → platelet activation + coagulation cascade
• Microthrombi in pulmonary capillaries, renal glomeruli, hepatic sinusoids → microvascular ischaemia
• Elevation of D-dimer (fibrin degradation product) correlates with severity and mortality
• COVID-19-associated coagulopathy resembles but differs from classic DIC: thrombotic tendency predominates over bleeding
Laboratory findings in COVID-19 (must memorise):
| Parameter | Finding in severe COVID-19 | Mechanism |
|---|---|---|
| Lymphocyte count | ↓↓ Lymphopenia | Direct viral infection of T-cells; lymphocyte apoptosis driven by cytokines |
| CRP | ↑↑ | Hepatic acute-phase response driven by IL-6 |
| Ferritin | ↑↑ | Released from macrophages (hyperactivated); marker of macrophage activation syndrome |
| D-dimer | ↑↑ | Microthrombus formation + fibrinolysis |
| LDH | ↑ | Tissue/cell damage (lung, liver, myocardium) |
| Procalcitonin | Normal/mildly ↑ (unless bacterial superinfection) | Helpful to rule out bacterial co-infection |
| RT-PCR (SARS-CoV-2) | Positive (nasopharyngeal swab) | Detection of viral RNA — gold standard for diagnosis |
Autopsy findings in COVID-19 deaths:
• Heavy, congested lungs (2–3× normal weight) — DAD + oedema
• Diffuse microthrombi in pulmonary vasculature
• Type II pneumocyte hyperplasia and hyaline membranes
• Multinucleated syncytial cells (viral cytopathic effect)
• Extrapulmonary: myocarditis (lymphocytic), acute tubular injury (kidney), hepatocyte necrosis (liver)
SELF-CHECK
An autopsy on a 55-year-old who died of severe COVID-19 shows bilateral heavy lungs with eosinophilic hyaline membranes lining alveolar walls, type II pneumocyte hyperplasia, and microthrombi in pulmonary capillaries. Ante-mortem bloods showed lymphopenia and markedly elevated D-dimer. Which pathological process is PRIMARILY responsible for the hyaline membranes, and which single laboratory marker BEST reflects the degree of coagulopathy?
A. Granulomatous inflammation; CRP best reflects coagulopathy
B. Neutrophilic exudation forming purulent fibrin; procalcitonin reflects coagulopathy
C. Diffuse alveolar damage (exudative phase) — protein-rich oedema fluid + fibrin precipitate as hyaline membranes on alveolar surfaces; D-dimer best reflects coagulopathy
D. Organising pneumonia (proliferative phase); LDH reflects coagulopathy
Reveal Answer
Answer: C. Diffuse alveolar damage (exudative phase) — protein-rich oedema fluid + fibrin precipitate as hyaline membranes on alveolar surfaces; D-dimer best reflects coagulopathy
Hyaline membranes are the histological signature of the exudative phase of Diffuse Alveolar Damage (DAD). When the alveolar–capillary barrier is disrupted (here by SARS-CoV-2 cytopathic injury + cytokine-driven endothelial damage), protein-rich oedema fluid and fibrin flood the alveolar space and precipitate along alveolar walls as eosinophilic 'glassy' membranes. D-dimer is the best marker of coagulopathy in COVID-19: it reflects fibrin formation and lysis (microthrombus turnover) and correlates strongly with severity and in-hospital mortality. CRP reflects inflammation; LDH reflects tissue damage; procalcitonin reflects bacterial superinfection.