Page 5 of 28
PA23.3-4 | Peptic Ulcer Disease & Gastric Carcinoma — SDL Guide
Learning Objectives
- Distinguish acute from chronic gastritis and identify the roles of H. pylori and autoimmune mechanisms.
- Explain peptic ulcer disease as an imbalance between aggressive and defensive mucosal factors.
- Describe the gross and microscopic features of benign versus malignant gastric ulcers.
- Outline the four histological zones of a chronic peptic ulcer.
- Enumerate the complications of peptic ulcer disease.
- Classify gastric carcinoma using the Lauren classification and describe the features of intestinal versus diffuse type.
- Explain the pathogenesis of gastric carcinoma from H. pylori infection to carcinoma.
- Identify the routes of metastatic spread of gastric carcinoma and their eponymous designations.
INSTRUCTIONS
Peptic ulcer disease and gastric carcinoma together account for enormous morbidity and mortality worldwide, and both are intimately linked to H. pylori infection. Understanding how the stomach's defensive mechanisms fail — and how chronic inflammation can progress to malignancy — is essential for clinical diagnosis, rational pharmacotherapy, and recognising alarm symptoms that demand early endoscopy. This module builds directly on your Year-1 physiology of gastric secretion and your microbiology knowledge of H. pylori.
References
- Robbins & Kumar Basic Pathology, 11th ed., Ch 14 — Gastrointestinal Tract (textbook)
- Harsh Mohan Textbook of Pathology, 8th ed., Ch 19 — Stomach (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 52-year-old farmer presents with a 3-month history of epigastric pain that wakes him at 2 AM, relieved by milk. Endoscopy reveals a 1.2 cm punched-out ulcer in the duodenal bulb with clean, sloping edges. Biopsy of the antrum: dense neutrophilic infiltrate and curved gram-negative rods. His neighbour — same village, similar diet — had an endoscopy last month: a 3.5 cm ulcer in the gastric body with heaped-up, irregular margins and necrotic base. One is benign; one demands urgent surgery. What determines the difference, and how does the stomach move from ulcer to cancer?
WHY THIS MATTERS
Peptic ulcer disease affects roughly 10% of the global population at some point in their lives; H. pylori infects over half of humanity. Gastric carcinoma kills nearly 800,000 people annually — the third leading cause of cancer death worldwide — yet it is largely preventable. As a clinician you will prescribe proton-pump inhibitors, interpret endoscopy reports, and decide when an 'ulcer' needs urgent biopsy. As a pathologist you will distinguish benign from malignant ulcers on gross and microscopic grounds, classify carcinomas, and guide surgical margins. This knowledge is examined in PG entrance tests, USMLE-style integrated questions, and — most importantly — in clinical practice from Day 1 of your posting.
RECALL
Before proceeding, activate your prior knowledge:
- From Physiology (Year 1): What stimulates gastric acid secretion (G cells → gastrin, parietal cells → HCl)? What is the role of prostaglandins in mucosal protection?
- From Microbiology: What is the Gram staining and urease activity of H. pylori? Why is the CLO (Campylobacter-like organism) test useful?
- From Pharmacology: How do proton-pump inhibitors differ from H2-blockers in mechanism?
- From PA SDL 1 (Gastritis overview): What is the distinction between acute erosive gastritis and chronic gastritis?
If any of these feel hazy, spend 3 minutes reviewing before proceeding — this module builds on all four.
Gastritis — Brief Overview (Context for PUD)
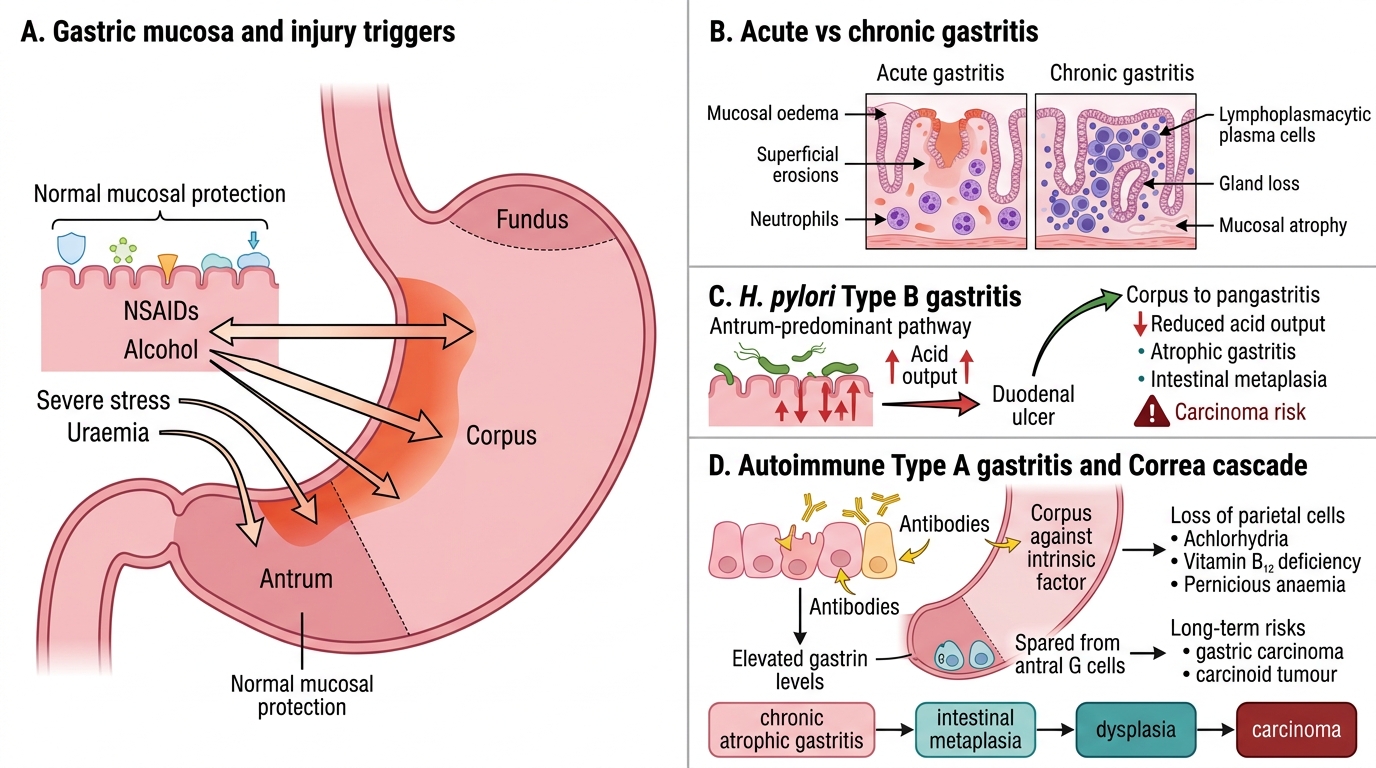
Gastritis: Types, Mechanisms, and Clinical Consequences
Gastritis is inflammation of the gastric mucosa. Understanding its two main forms contextualises peptic ulcer disease.
Acute gastritis is characterised by transient neutrophilic infiltration, mucosal oedema, and superficial erosions. Common causes include NSAIDs (inhibit COX-1 → reduced prostaglandin synthesis → loss of mucosal protection), alcohol, severe stress (stress ulcers in ICU patients), and uraemia. It is usually self-limiting.
Chronic gastritis involves persistent lymphoplasmacytic infiltration and eventual mucosal atrophy. Two major types:
- H. pylori gastritis (Type B) — the commonest form worldwide. Affects the antrum predominantly. Leads to antral-predominant acid hypersecretion → duodenal ulcers (ulcer-prone phenotype). If it spreads to the corpus (pangastritis), acid output falls → atrophic gastritis → intestinal metaplasia → cancer risk.
- Autoimmune gastritis (Type A) — antibodies against parietal cells and intrinsic factor. Affects the corpus/fundus. Causes achlorhydria, vitamin B12 deficiency (pernicious anaemia), and elevated gastrin (antral G cells are spared but respond to the achlorhydria). Long-term risk of gastric carcinoma and carcinoid tumours.
The transition from chronic atrophic gastritis → intestinal metaplasia → dysplasia → carcinoma (Correa's cascade) is central to understanding gastric cancer pathogenesis.
Peptic Ulcer Disease — Definition & Epidemiology
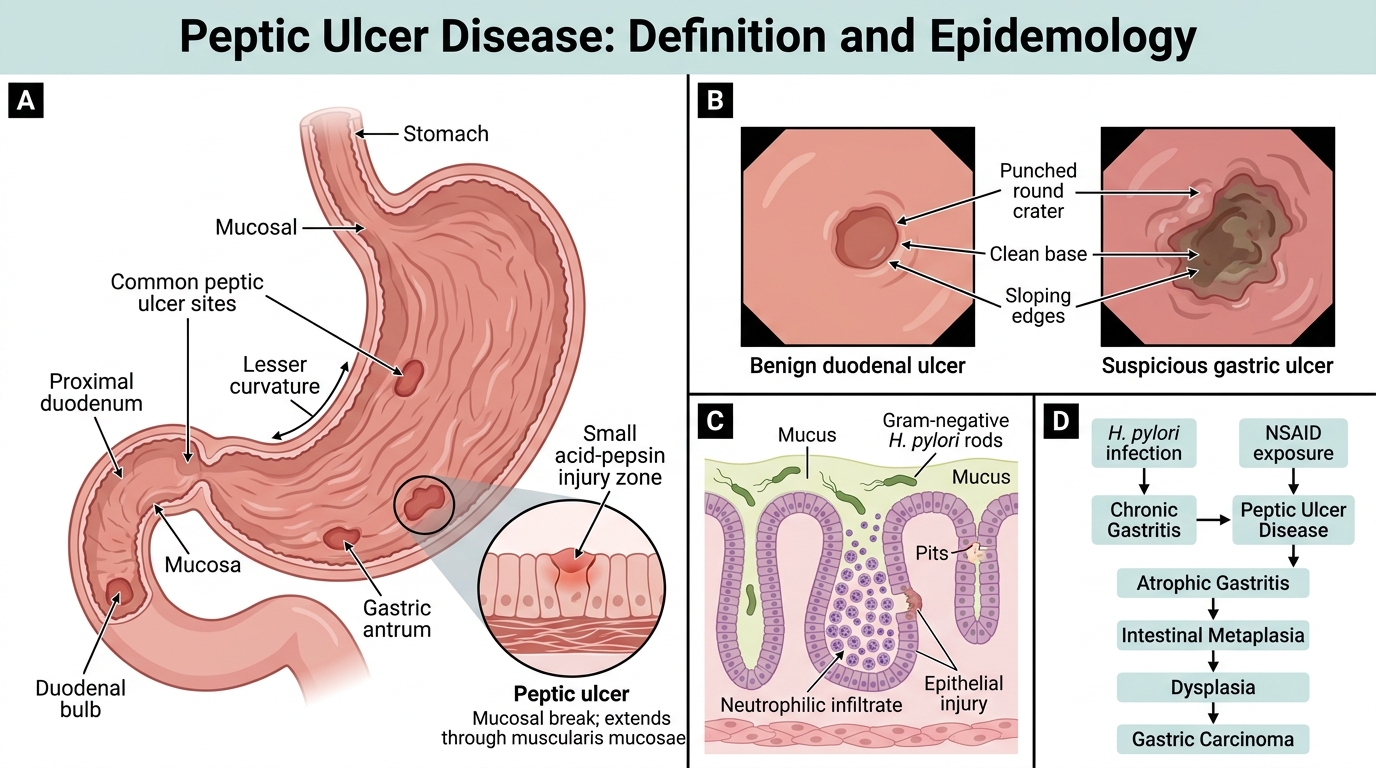
Peptic Ulcer Disease: Definition, Sites, and Clinical Significance
Peptic ulcer disease (PUD) refers to discontinuous mucosal defects that extend through the muscularis mucosae, caused by the action of acid and pepsin. By definition, they penetrate deeper than erosions (which are superficial).
Sites: 95% occur in the duodenum (first part, anterior wall) or stomach (lesser curvature, antrum/body junction). The oesophagus (Barrett's) and Meckel's diverticulum are rarer sites.
Epidemiology:
- Duodenal ulcers (DU): more common (4:1 over gastric ulcers), peak age 30-55, men > women, virtually always benign.
- Gastric ulcers (GU): peak age 50-70, male predominance; small risk of malignancy (sample all gastric ulcers).
- H. pylori is found in 90-95% of DU and 70-80% of GU.
- NSAID use accounts for most H. pylori-negative ulcers.
Pathogenesis — Aggressive vs Defensive Factor Imbalance
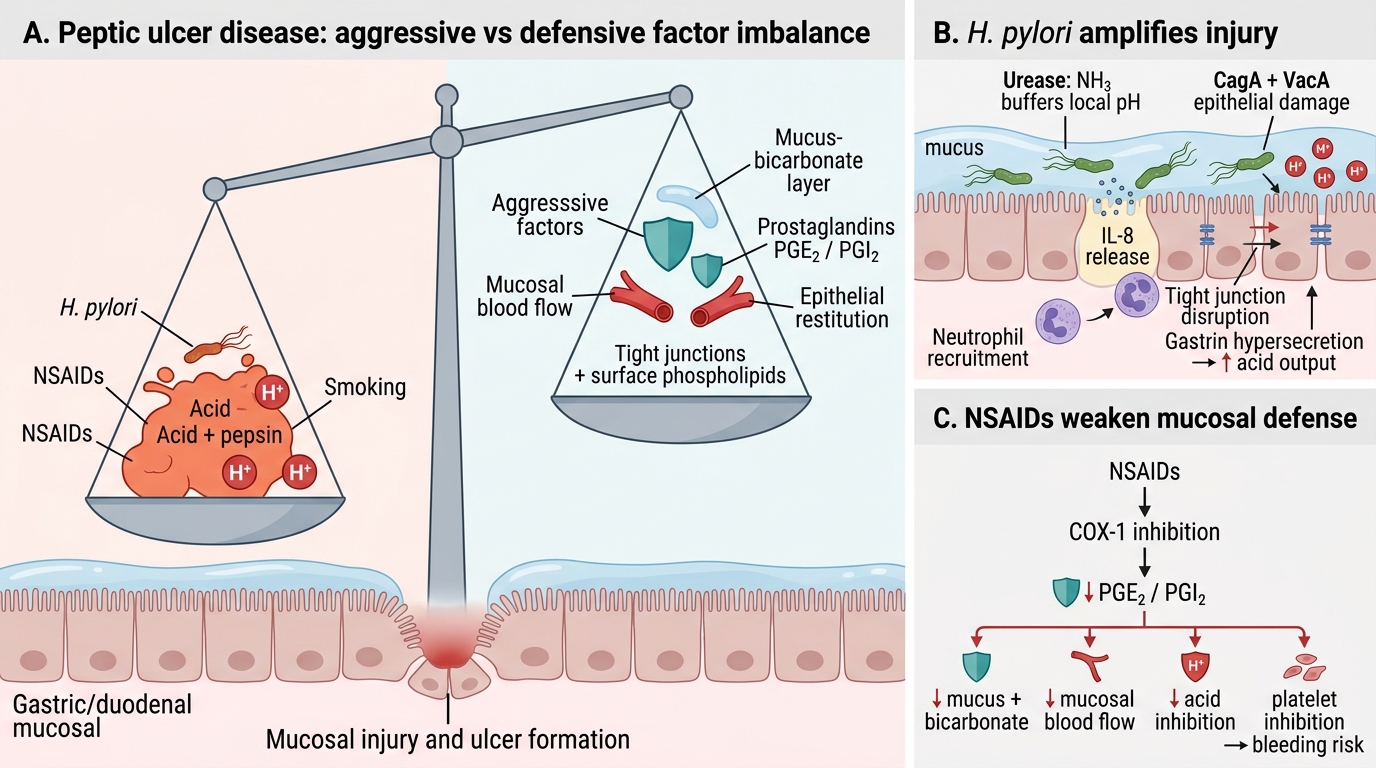
Pathogenesis of Peptic Ulcer Disease: Aggressive vs Defensive Imbalance
PUD results from an imbalance tilting toward mucosal injury. Memorise this as a balance model:
Aggressive factors (injurious):
• H. pylori — colonises mucus layer; produces urease (NH3 → neutralises local pH, allows survival); secretes CagA (cytotoxin-associated gene A) and VacA (vacuolating cytotoxin A) → epithelial damage, IL-8 release, neutrophil recruitment; disrupts tight junctions; promotes gastrin hypersecretion (increased acid output).
• NSAIDs — inhibit COX-1 → decreased mucosal prostaglandin (PGE2, PGI2) → reduced mucus/bicarbonate secretion, reduced mucosal blood flow, enhanced platelet aggregation inhibition (bleeding risk).
• Acid and pepsin — the final effectors of mucosal digestion; essential co-factors (ulcers don't form in achlorhydria).
• Smoking — reduces bicarbonate, impairs mucosal blood flow, delays healing.
Defensive factors (protective):
• Mucus-bicarbonate layer — mucus gel traps bicarbonate, neutralises luminal H⁺ before it contacts epithelium.
• Prostaglandins (PGE2, PGI2) — stimulate mucus + bicarbonate secretion, promote mucosal blood flow, inhibit parietal cell acid secretion.
• Mucosal blood flow — delivers oxygen + nutrients, removes back-diffused H⁺.
• Epithelial restitution — rapid migration of cells to re-surface small erosions.
• Tight junctions and surface phospholipids — hydrophobic barrier.
H. pylori primarily attacks defensive mechanisms (mucus disruption) AND amplifies aggression (gastrin → acid). NSAIDs primarily knock out prostaglandins.
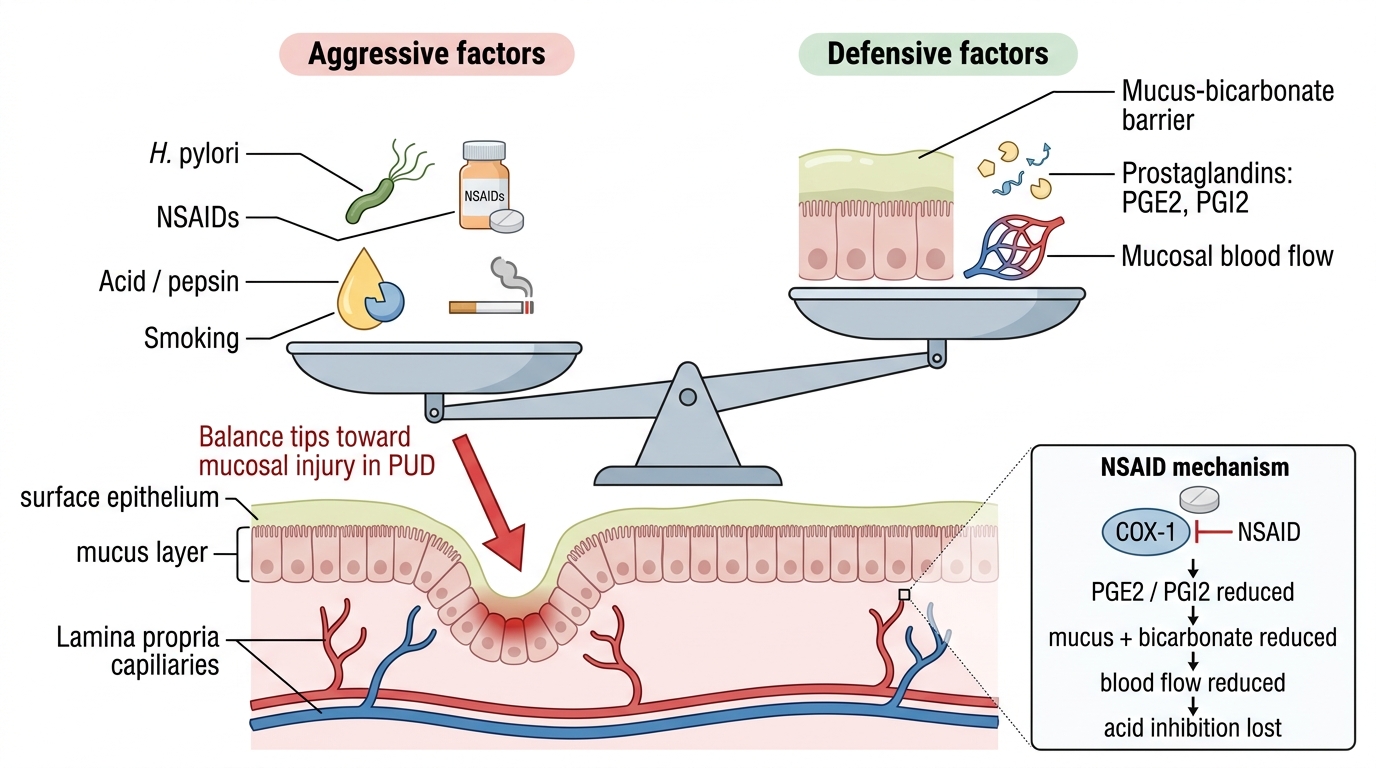
Aggressive and Defensive Factors in Peptic Ulcer Disease
SELF-CHECK
A patient on long-term ibuprofen for rheumatoid arthritis develops a gastric ulcer. The PRIMARY mechanism by which NSAIDs cause peptic ulceration is:
A. Direct toxic effect of the drug on the gastric epithelium after systemic absorption
B. Inhibition of COX-1 leading to reduced prostaglandin synthesis and loss of mucosal cytoprotection
C. Stimulation of gastrin secretion from antral G cells
D. Suppression of the immune response allowing H. pylori colonisation
Reveal Answer
Answer: B. Inhibition of COX-1 leading to reduced prostaglandin synthesis and loss of mucosal cytoprotection
NSAIDs inhibit COX-1 (constitutively expressed), reducing synthesis of PGE2 and PGI2. These prostaglandins normally stimulate mucus and bicarbonate secretion, maintain mucosal blood flow, and inhibit acid secretion. Their loss destroys the defensive barrier. Topical irritation exists but is secondary; the systemic effect via PG depletion is the dominant mechanism — which is why parenteral/enteric-coated NSAIDs also cause ulcers.