Page 2 of 25
PA20.1 | Normal Haemostasis — SDL Guide (Part 2)
Secondary Haemostasis: The Coagulation Cascade
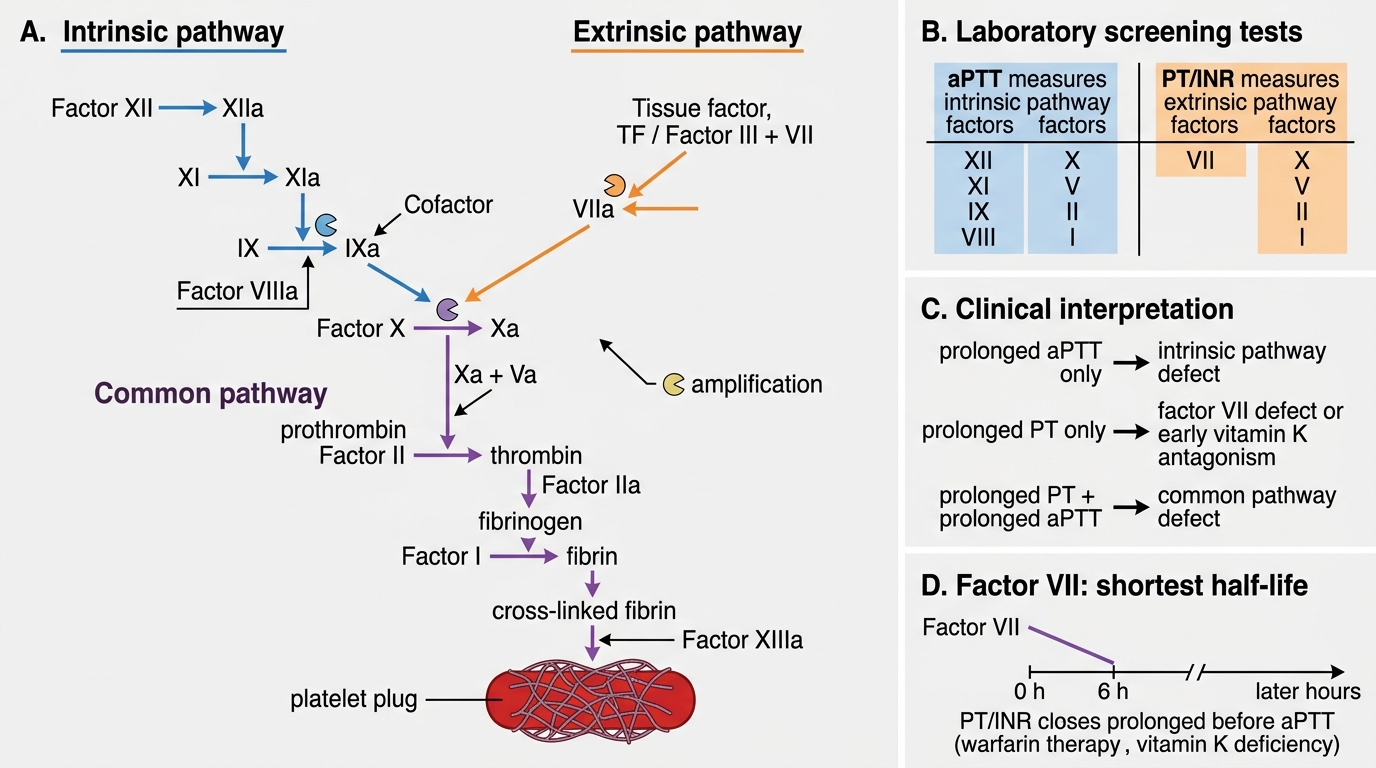
Secondary Haemostasis: Coagulation Cascade
The coagulation cascade converts the loose platelet plug into a stable, cross-linked fibrin clot. It is a series of enzymatic reactions where each activated factor (zymogen → serine protease) activates the next, producing enormous amplification.
Classically divided into three pathways:
Intrinsic pathway (contact activation):
XII → XI → IX; VIII is a cofactor for IXa
All factors in this pathway are measured by aPTT (activated partial thromboplastin time). Prolonged aPTT = defect in XII, XI, IX, or VIII.
Extrinsic pathway (tissue factor pathway):
Tissue factor (TF, aka Factor III) + VII → VIIa
This is the physiologically dominant initiating pathway. Measured by PT/INR (prothrombin time). Prolonged PT = defect in VII, or in the common pathway.
Common pathway:
X + Va (cofactor) → prothrombin (II) → thrombin → fibrinogen (I) → fibrin → cross-linked fibrin (XIII)
Affected by defects in X, V, II, I, or XIII. Prolonged PT + prolonged aPTT = common pathway defect.
IMPORTANT: Factor VII has the shortest half-life (~6 hours). Therefore, warfarin or vitamin K deficiency first prolongs the PT (extrinsic arm), before the aPTT.

Complete Coagulation Cascade: Classical Pathways, Thrombin Function, and Cell-Based Model
Thrombin: The Central Molecule of Haemostasis
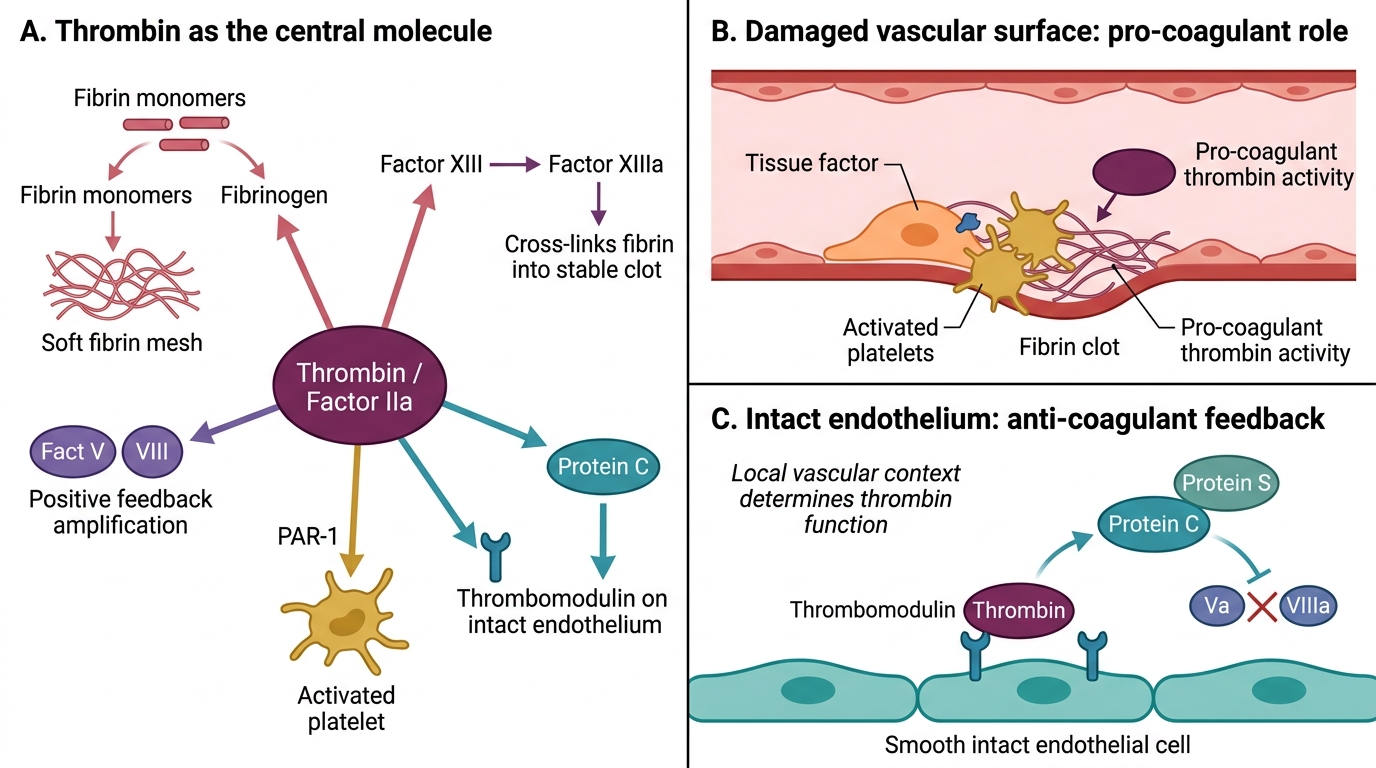
Thrombin: Central Regulator of Haemostasis
Thrombin (activated factor IIa) is the pivotal effector of the coagulation cascade. It is not merely a fibrinogen-cleaver — it is a master amplifier:
- Cleaves fibrinogen → fibrin monomers → polymerisation → soft clot
- Activates factor XIII → cross-links fibrin → insoluble, stable clot
- Activates factors V and VIII (positive feedback amplification of its own generation)
- Activates platelets (via PAR-1 receptor) — the most potent platelet activator
- Binds thrombomodulin on intact endothelium → activates Protein C (anticoagulant feedback)
Thrombin is simultaneously pro-coagulant (on damaged surfaces) and anti-coagulant (on intact endothelium). The local vascular context determines which role predominates.
The Cell-Based Model: How Coagulation Actually Works In Vivo

Cell-Based Model of Coagulation In Vivo
The classical cascade model (intrinsic vs. extrinsic pathways) is a lab construct, not a physiological reality. The modern cell-based model better explains why haemophilia A (factor VIII deficiency) causes severe bleeding even though factor VIII is 'only' in the intrinsic pathway.
The cell-based model has three phases:
1. Initiation — on tissue factor–bearing cells (fibroblasts, monocytes exposed at injury site):
TF + VIIa → small amounts of Xa, IIa (thrombin) — not enough for clot, but enough to prime platelets
2. Amplification — on activated platelets:
Thrombin activates VIII (from vWF–VIII complex), V, and XI on the platelet surface; platelet surface becomes a phospholipid scaffold
3. Propagation — on platelets:
IXa–VIIIa complex (tenase) + Xa–Va complex (prothrombinase) → explosive thrombin burst → fibrin clot
Clinical insight: Without factor VIII (haemophilia A), the tenase complex cannot form on the platelet surface during propagation → insufficient thrombin burst → clot forms but is fragile → deep tissue bleeding. The extrinsic pathway initiates normally (PT is normal), but the intrinsic/amplification arm collapses (aPTT is prolonged).
SELF-CHECK
In the cell-based model of coagulation, which phase generates the explosive thrombin burst sufficient to polymerise fibrin?
A. Initiation on tissue factor–bearing cells
B. Contact activation by factor XII
C. Amplification on damaged endothelium
D. Propagation on activated platelet surfaces
Reveal Answer
Answer: D. Propagation on activated platelet surfaces
Propagation — occurring on activated platelet phospholipid surfaces via the tenase (IXa–VIIIa) and prothrombinase (Xa–Va) complexes — generates the explosive thrombin burst required for stable fibrin polymerisation. Initiation on TF-bearing cells produces only a small priming amount of thrombin. Factor XII contact activation is not required in vivo (XII deficiency does not cause clinical bleeding). Endothelium is anti-thrombotic.