Page 3 of 25
PA20.1 | Normal Haemostasis — SDL Guide (Part 3)
Fibrinolysis: Dissolving the Clot

Fibrinolytic Pathway and Regulation
Once wound healing is underway, the fibrin clot must be dissolved. The fibrinolytic system prevents excessive clotting and restores vessel patency.
Key pathway:
Plasminogen (inactive) → plasmin (active serine protease)
Activators:
• tPA (tissue plasminogen activator) — released by endothelial cells; the therapeutic principle behind alteplase in acute MI/stroke
• uPA (urokinase-type plasminogen activator)
Plasmin cleaves fibrin → fibrin degradation products (FDPs) and D-dimer (a cross-link-specific FDP)
D-dimer is clinically important: its elevation confirms active fibrinolysis (clot formation AND dissolution). It is used as a sensitive screening test for DVT/PE — but it is non-specific (elevated in many inflammatory states).
Inhibitors of fibrinolysis:
• PAI-1 (plasminogen activator inhibitor-1): inhibits tPA/uPA
• α₂-antiplasmin: directly inhibits free plasmin

Fibrinolytic Pathway and Regulation
Natural Anticoagulant Mechanisms
⚑ AI image — pending faculty review (auto-QA score 6/10; best of 3 attempts)
Natural Anticoagulant Mechanisms
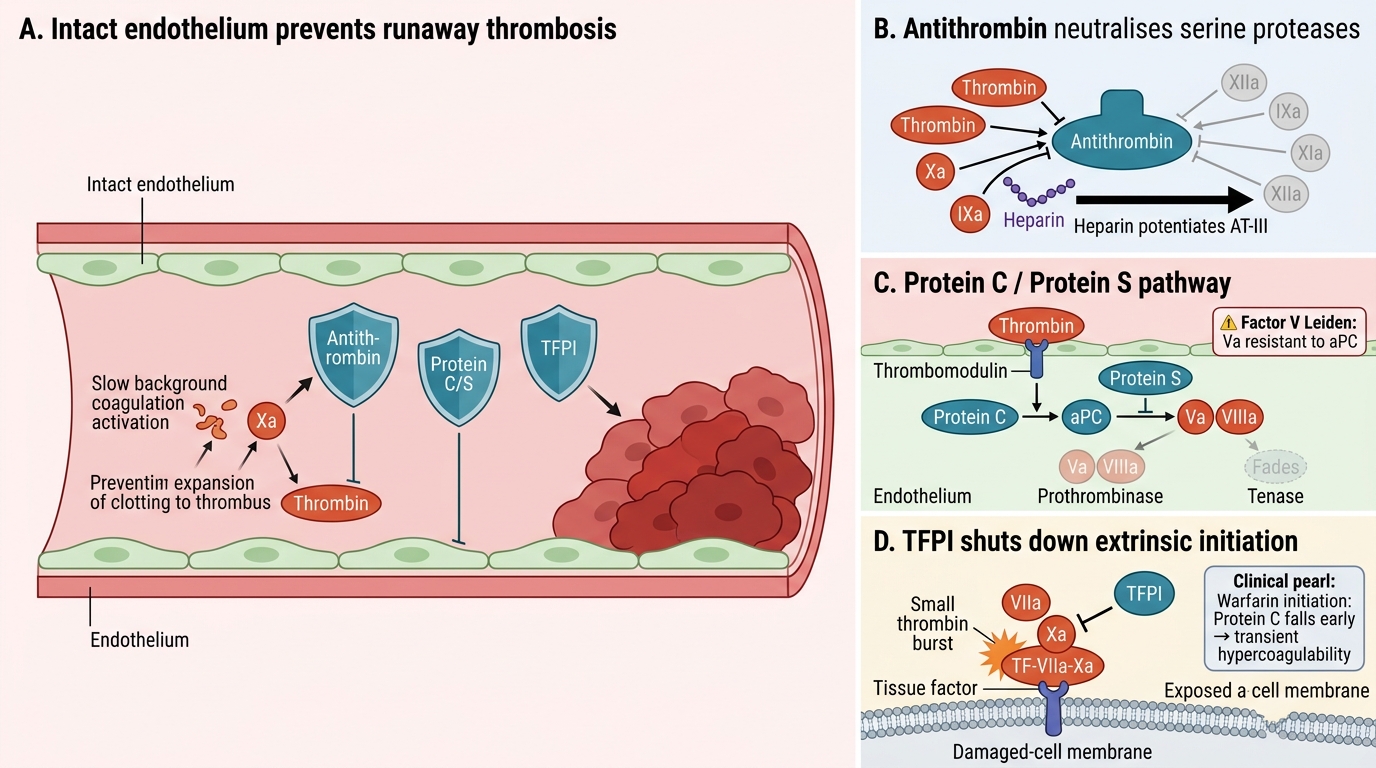
Coagulation is continuously activated at low levels. The natural anticoagulant system prevents runaway thrombosis in intact vessels. Three major mechanisms:
1. Antithrombin (AT-III)
A serine protease inhibitor (serpin) that neutralises thrombin, Xa, IXa, XIa, XIIa. Its activity is dramatically potentiated by heparin — the therapeutic basis of unfractionated and low-molecular-weight heparin. Antithrombin deficiency → thrombophilia.
2. Protein C / Protein S pathway
• Thrombin binds thrombomodulin on intact endothelium → activates Protein C
• Activated Protein C (aPC) + Protein S (cofactor) → degrades Va and VIIIa → shuts down the prothrombinase and tenase complexes
• Factor V Leiden (Arg506Gln mutation) renders Va resistant to aPC → most common inherited thrombophilia (5% of Europeans)
• Protein C and S are vitamin K–dependent: warfarin depletes them before depleting pro-coagulant factors, paradoxically causing a brief hypercoagulable state at warfarin initiation
3. Tissue Factor Pathway Inhibitor (TFPI)
Produced by endothelial cells; directly inhibits the TF–VIIa–Xa complex, shutting down extrinsic pathway initiation after the first small burst of thrombin is generated. This is why the extrinsic pathway alone cannot sustain a clot — the cell-based amplification/propagation phases (intrinsic arm) are essential.
CLINICAL PEARL
Warfarin paradox at initiation: Protein C has the shortest half-life among vitamin K–dependent proteins (~8 hours). When warfarin is started, Protein C is depleted before the pro-coagulant factors II, IX, and X. This transiently tips the balance toward thrombosis — the mechanism of warfarin-induced skin necrosis, which occurs in protein C–deficient patients given warfarin without heparin bridging. This is why heparin overlap for 4–5 days is mandatory when starting warfarin therapy.
Vitamin K–Dependent Clotting Factors

Vitamin K–Dependent Clotting Factors
Vitamin K (fat-soluble) is a cofactor for the enzyme that adds γ-carboxyglutamate (Gla) residues to clotting factors. These Gla residues allow the factors to bind calcium and assemble on phospholipid membranes — essential for their activity.
Vitamin K–dependent pro-coagulant factors: II (prothrombin), VII, IX, X — mnemonic: '1972' (I=1, reversed; 9, 7, 2)
Vitamin K–dependent anticoagulant proteins: Protein C, Protein S
Clinical implications:
• Warfarin blocks vitamin K epoxide reductase (VKOR), depleting all six proteins above
• Vitamin K deficiency (malabsorption, prolonged antibiotics, newborn — physiological) → depletes all six → prolonged PT and aPTT
• Neonatal haemorrhagic disease (vitamin K deficiency of the newborn) — prevented by prophylactic IM vitamin K at birth, now standard of care
• VKORC1 and CYP2C9 polymorphisms explain the enormous inter-individual variation in warfarin dosing