Page 1 of 13
PA17.1 | Aplastic Anaemia & Bone Marrow Examination — SDL Guide
Learning Objectives
- Define aplastic anaemia and distinguish it from other causes of pancytopenia
- Enumerate the aetiology of aplastic anaemia including idiopathic, acquired, and inherited causes
- Explain the immune-mediated and intrinsic stem-cell pathogenesis of aplastic anaemia
- Describe the peripheral blood and bone marrow findings in aplastic anaemia
- Apply the Camitta severity criteria to classify disease severity
- Enumerate the indications for bone marrow aspiration and biopsy
- Compare the information obtained from bone marrow aspirate versus trephine biopsy
- Interpret the significance of a 'dry tap' and estimate marrow cellularity from a trephine biopsy
INSTRUCTIONS
Aplastic anaemia is a life-threatening marrow failure syndrome where the stem-cell pool is destroyed, leaving all three blood lineages depleted. Understanding its aetiology, immune pathogenesis, and diagnostic work-up — including bone marrow aspiration and biopsy — is a core Year-2 Pathology competency (PA17.1) that integrates directly with haematology clinical postings. This module builds on Year-1 cell-biology and haematopoiesis, and sets the foundation for recognising and managing marrow failure in clinical practice.
References
- Robbins & Kumar: Basic Pathology, 11th ed., Ch 13 (Red Cell Disorders) (textbook)
- Harsh Mohan: Textbook of Pathology, 8th ed., Ch 12 (textbook)
- Dacie & Lewis: Practical Haematology, 12th ed. (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 19-year-old engineering student presents with progressive fatigue over two months, five episodes of epistaxis in one week, and three consecutive febrile illnesses — each treated with a different antibiotic by a local clinic. Examination: pallor, petechiae on both legs, NO splenomegaly, NO lymphadenopathy. CBC: Hb 5.8 g/dL, TLC 1,400/µL (neutrophils 400/µL), platelets 14,000/µL. Peripheral smear: normocytic-normochromic RBCs, no blasts, no hypersegmented neutrophils. What single bone marrow finding would clinch the diagnosis — and why does the absence of organomegaly matter?
WHY THIS MATTERS
Aplastic anaemia is the most dramatic presentation of marrow failure you will encounter. Because it mimics leukaemia clinically but demands a completely different management pathway — haematopoietic stem-cell transplant rather than chemotherapy — misdiagnosis is catastrophic. Bone marrow examination is the gatekeeper: a Year-2 graduate must know when to request it, what technique to use, and how to interpret the report. These skills carry directly into medicine, paediatrics, and oncology postings.
RECALL
Before proceeding, activate what you already know:
- From Year-1 Physiology — where are blood cells produced in an adult, and what is the approximate cellularity of normal adult marrow?
- From Year-1 Biochemistry — what is the role of CD34+ haematopoietic stem cells?
- From Year-1 Pathology — define pancytopenia and list two causes you have already encountered (e.g., megaloblastic anaemia, hypersplenism).
Keep these in mind — they will form the baseline against which aplastic anaemia's abnormality becomes sharply defined.
Definition of Aplastic Anaemia

Definition of Aplastic Anaemia
Aplastic anaemia is defined as pancytopenia accompanied by a hypocellular bone marrow in the absence of:
- Abnormal (dysplastic or malignant) cells
- Marrow fibrosis
- Marrow infiltration (e.g., by granulomas, metastatic deposits)
All three exclusions are diagnostically critical. Pancytopenia alone is non-specific; the marrow morphology differentiates aplastic anaemia from myelodysplastic syndrome (MDS), leukaemia, and myelofibrosis.
Pancytopenia = simultaneous reduction in erythrocytes, leucocytes (especially neutrophils), and platelets — producing anaemia, neutropenic infections, and thrombocytopenic bleeding respectively.
The marrow is replaced by fat cells and stroma; haematopoietic precursors are severely depleted or absent.
Aetiology
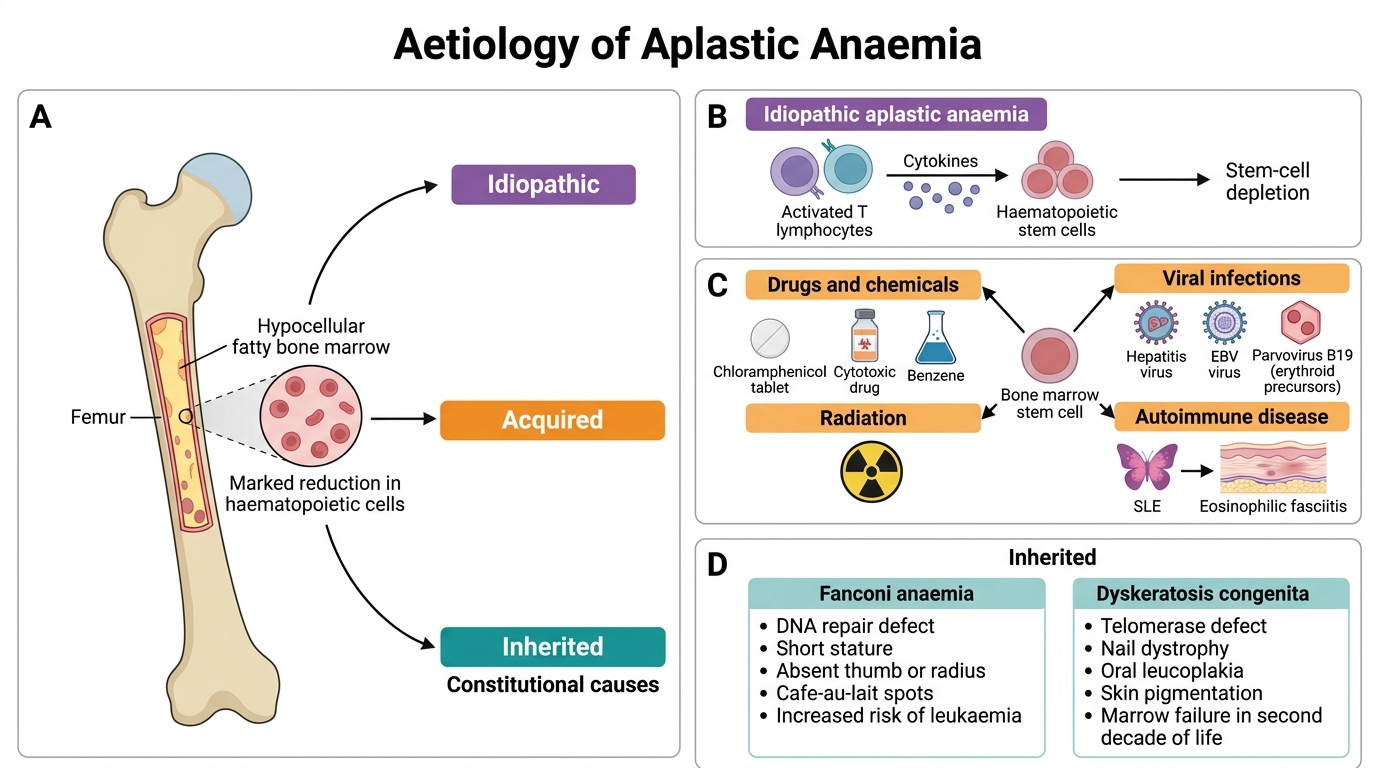
Aetiology of Aplastic Anaemia
Aplastic anaemia is classified by cause:
1. Idiopathic (~70% of cases)
No identifiable cause found; presumed autoimmune in most cases.
2. Acquired — secondary causes
Drugs and chemicals (most important acquired group):
- Chloramphenicol — classic dose-independent idiosyncratic marrow suppression (also has predictable dose-dependent suppression)
- Cytotoxic drugs (busulfan, cyclophosphamide) — dose-dependent, predictable
- Benzene — occupational exposure; direct stem-cell toxin
- Radiation — ionising radiation destroys dividing stem cells
Viral infections:
- Hepatitis (non-A, non-B, non-C seronegative hepatitis) — post-hepatitis aplasia, often severe
- Epstein-Barr virus (EBV) — rare; usually mild and transient
- Parvovirus B19 — selectively destroys erythroid precursors → pure red cell aplasia (not full aplastic anaemia; note the distinction)
Autoimmune diseases:
- Systemic lupus erythematosus (SLE), eosinophilic fasciitis
3. Inherited (Constitutional) Aplastic Anaemia
- Fanconi anaemia — autosomal recessive; DNA repair defect (FANC genes); associated with skeletal anomalies (absent thumbs/radii), café-au-lait spots, short stature; presents in childhood; high leukaemia risk
- Dyskeratosis congenita — X-linked (most common) or AR; telomerase gene mutations; triad: nail dystrophy, oral leucoplakia, skin pigmentation; marrow failure in second decade
Pathogenesis

Pathogenesis of Aplastic Anaemia
Two mechanisms operate, often together:
1. Immune-mediated T-cell destruction (dominant mechanism, ~80%)
Activated cytotoxic T lymphocytes (CD8+) attack haematopoietic stem cells through:
- Fas–FasL interaction (apoptosis pathway)
- Perforin/granzyme release
- Interferon-γ and TNF-α secretion → haematopoietic suppression
Evidence: most patients respond to immunosuppressive therapy (anti-thymocyte globulin + ciclosporin); lymphocytes infiltrate marrow; CD4:CD8 ratio is inverted in marrow.
The trigger is unknown for idiopathic cases; drugs/viruses likely break self-tolerance in genetically predisposed individuals.
2. Intrinsic stem-cell defect
Inherited cases (Fanconi anaemia) have a primary defect in DNA repair, causing stem cells to undergo apoptosis after replication stress. This mechanism also operates as a secondary pathway in acquired aplasia after repeated immune assault.
Key concept: In aplastic anaemia the marrow space is preserved but empty — unlike myelofibrosis where fibrosis physically replaces haematopoietic tissue, or leukaemia where blasts crowd out normal precursors.
CLINICAL PEARL
The NO-organomegaly rule: In aplastic anaemia there is NO hepatosplenomegaly and NO lymphadenopathy. This is a cardinal feature — and a key differential point. Splenomegaly suggests hypersplenism, portal hypertension, CML, or storage disorder. Hepatosplenomegaly + pancytopenia in a young adult should prompt Gaucher disease or hairy-cell leukaemia, not aplastic anaemia. If you find organomegaly, revise your diagnosis.