Page 10 of 32
PA24.{2,4} | Alcoholic Liver Disease, Cirrhosis & Hepatic Failure — SDL Guide
Learning Objectives
- Describe the three-stage spectrum of alcoholic liver disease: steatosis, alcoholic hepatitis, and cirrhosis — their reversibility, morphology, and pathogenesis via acetaldehyde and oxidative stress.
- Identify the histological hallmarks of alcoholic hepatitis: hepatocyte ballooning, Mallory-Denk bodies, and neutrophilic infiltrate.
- Define cirrhosis and distinguish micronodular from macronodular patterns; enumerate the major aetiologies.
- Explain the pathophysiology of acute (fulminant) and chronic hepatic failure, and correlate each clinical complication — jaundice, encephalopathy, coagulopathy, portal hypertension consequences, hepatorenal and hepatopulmonary syndromes — with its underlying mechanism.
- Briefly describe the Child-Pugh scoring concept and its clinical utility.
INSTRUCTIONS
The liver processes everything you eat, drink, and absorb — when it fails, every organ system feels the consequences. Alcohol is the most common toxic cause of chronic liver disease worldwide, and its three-stage spectrum illustrates how a reversible metabolic insult can burn through to permanent architectural destruction. Understanding this progression unlocks the mechanisms behind cirrhosis, portal hypertension, and hepatic failure — complications you will encounter in wards, surgery, and medicine clinics throughout your career. This module builds on Year-1 biochemistry (lipid metabolism, NADH/NAD+ ratio, oxidative stress) and hepatocyte ultrastructure from Anatomy.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch 18 — Liver and Biliary Tract (textbook)
- Harsh Mohan's Textbook of Pathology, 8th ed., Ch 19 — Pathology of the Liver (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 44-year-old man presents with a massively distended abdomen, yellow sclera, and confusion — he cannot say where he is. His wife says he has been drinking heavily for 15 years. Ultrasound shows a shrunken nodular liver with ascites. Coagulation studies return uncorrectable. His ammonia is 140 µmol/L.
What has gone wrong? And why does a damaged liver make a patient bleed, swell, and lose their mind — all at once?
WHY THIS MATTERS
Alcoholic liver disease is the leading cause of cirrhosis in most urban Indian hospitals. Hepatic failure — the end-stage consequence — carries a 30-day mortality of 50–90% in the fulminant form. Every complication you will manage in medicine and surgery rounds traces back to the pathological mechanisms covered here: failed synthetic function, altered haemodynamics, and toxic metabolite accumulation. This SDL also lays the foundation for SDL 4 (Portal Hypertension & Hepatic Tumours) and the clinical pharmacology of alcohol you will meet in Phase 3.
RECALL
Before continuing, confirm you can answer:
• What is the role of NADH/NAD+ ratio in hepatic lipid metabolism? (Year-1 Biochemistry)
• Which hepatic zone (zone 1/2/3) is most oxygen-poor and therefore most vulnerable to toxic injury? (Year-1 Anatomy)
• What is the difference between conjugated and unconjugated bilirubin, and which fraction rises first in hepatocellular disease? (Year-1 Physiology)
If uncertain, a quick review of those concepts now will make the mechanisms below significantly clearer.
Overview: The Three-Stage Spectrum of Alcoholic Liver Disease
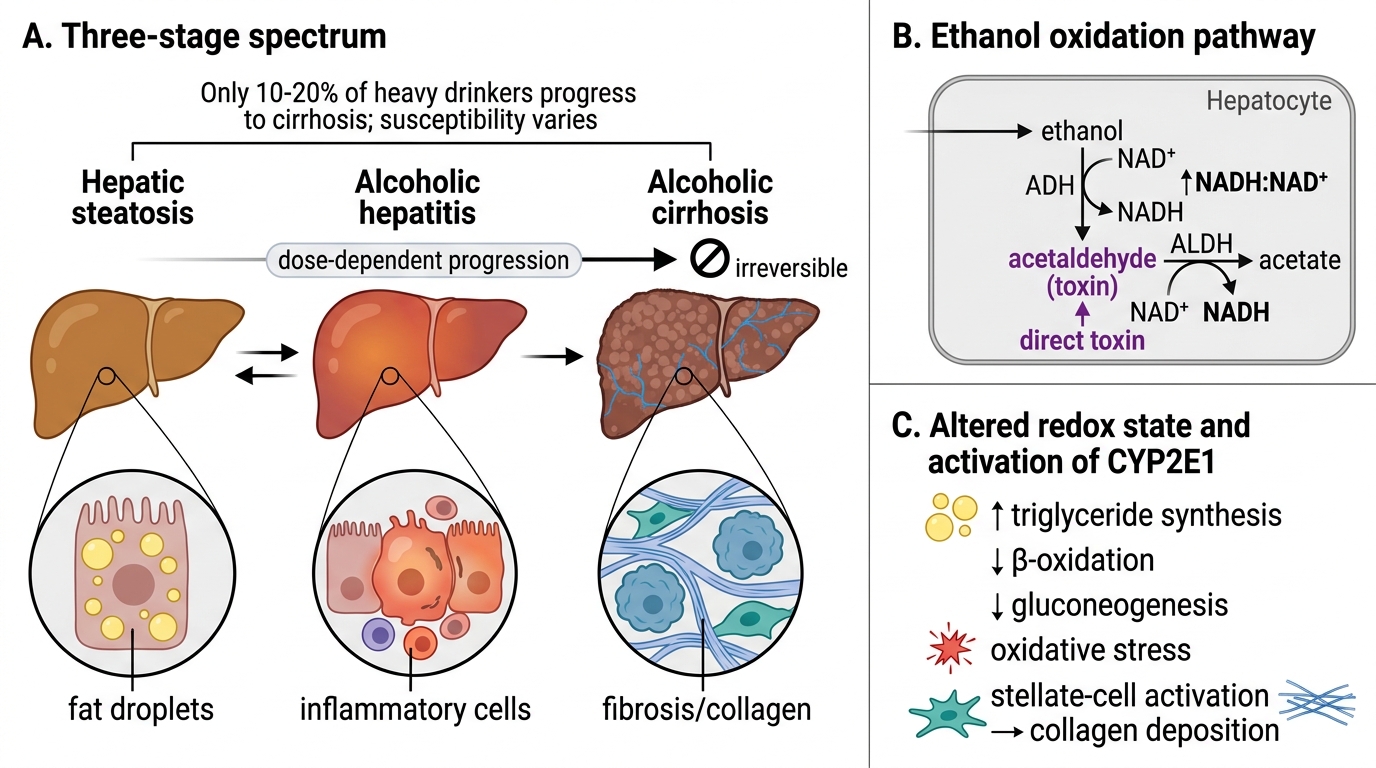
Alcoholic Liver Disease: Spectrum and Metabolic Basis
Alcohol (ethanol) causes liver injury in a predictable, dose-dependent three-stage progression:
- Hepatic steatosis (fatty liver) — reversible
- Alcoholic hepatitis — reversible if alcohol is stopped early, may progress
- Alcoholic cirrhosis — irreversible
The critical insight is that only 10–20% of heavy drinkers progress to cirrhosis; individual susceptibility (genetic, nutritional, co-infections) determines outcome. Each stage represents a quantitative increase in hepatocyte damage and a qualitative change in fibroblast/stellate-cell activation.
The key metabolic driver is ethanol oxidation via alcohol dehydrogenase (ADH) and ALDH, producing acetaldehyde (direct toxin) and NADH (↑NADH/NAD+ ratio). The shifted redox state inhibits β-oxidation of fatty acids, promotes triglyceride synthesis, and blocks gluconeogenesis — together these produce fat accumulation (steatosis).
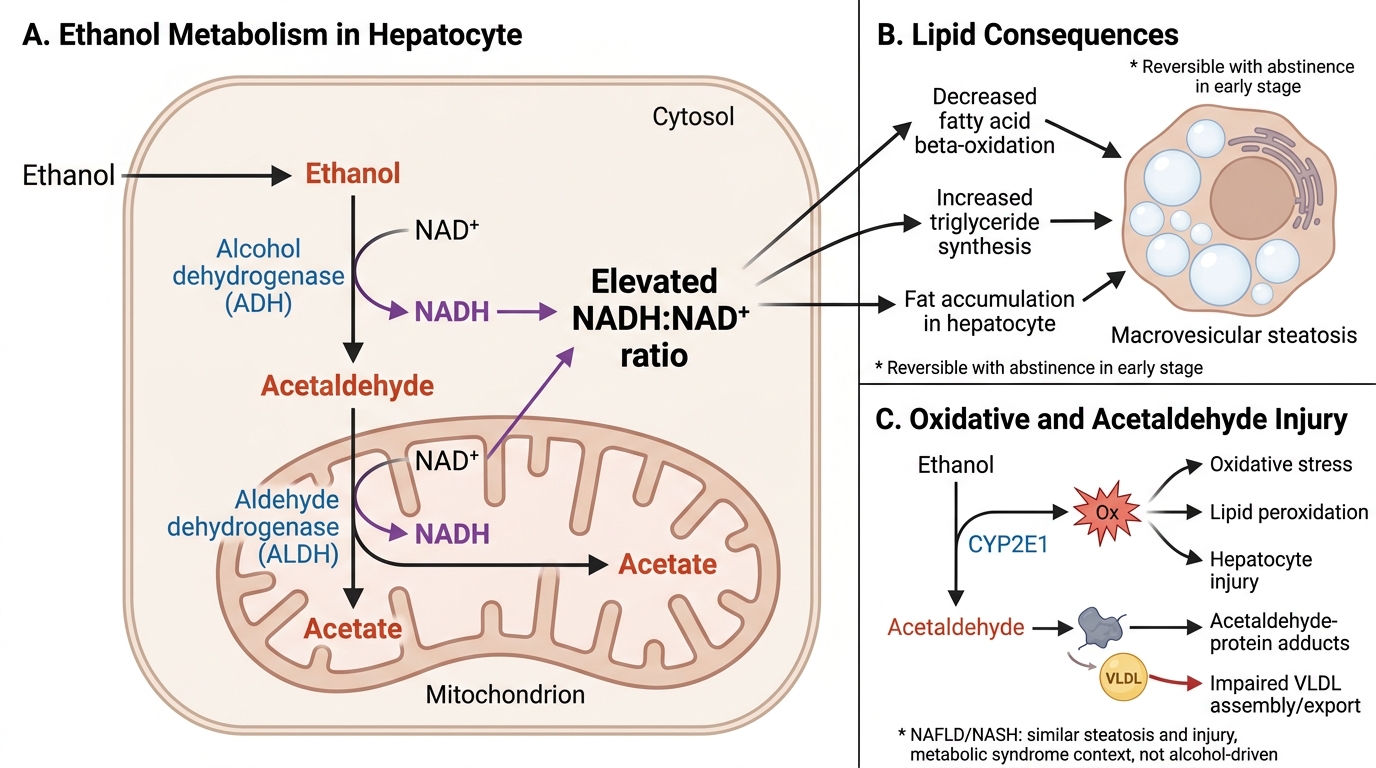
Ethanol Metabolism and Hepatic Steatosis Pathway
Note: NAFLD/NASH (non-alcoholic fatty liver disease / non-alcoholic steatohepatitis) mirrors alcoholic steatosis and hepatitis histologically but occurs in the context of metabolic syndrome (obesity, insulin resistance, type 2 diabetes) rather than alcohol. This underscores that steatosis and hepatocyte injury are final common pathways, not alcohol-specific responses.
Stage 1: Hepatic Steatosis (Alcoholic Fatty Liver)
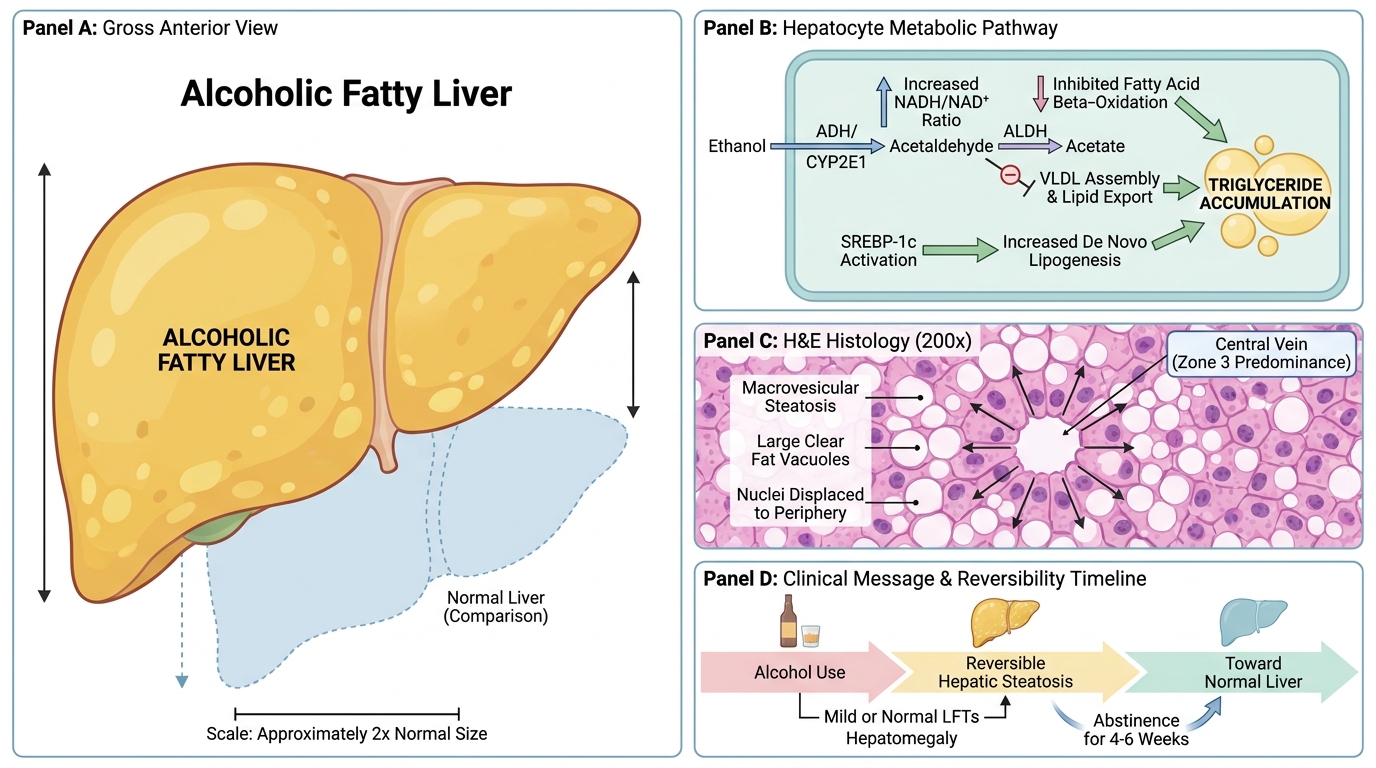
Alcoholic Hepatic Steatosis
Hepatic steatosis is the earliest, mildest, and fully reversible stage of alcoholic liver disease.
Pathogenesis — three converging mechanisms:
• ↑NADH/NAD+ ratio → inhibits fatty acid β-oxidation → fatty acids accumulate in hepatocytes
• Acetaldehyde directly disrupts lipid export (impairs VLDL assembly)
• ↑De novo lipogenesis via upregulated SREBP-1c
Gross morphology: Liver is enlarged (hepatomegaly), yellow, greasy, soft. May reach 4–6 kg.
Microscopy:
• Macrovesicular steatosis — large fat vacuoles pushing the nucleus to the periphery ("signet-ring" appearance)
• Predominantly zone 3 (centrilobular) distribution — highest alcohol/oxygen exposure
• No significant inflammation or fibrosis at this stage
Clinical: Usually asymptomatic. Hepatomegaly on examination. LFTs mildly elevated (or normal). Completely reversible with abstinence within 4–6 weeks — this is the key clinical message.
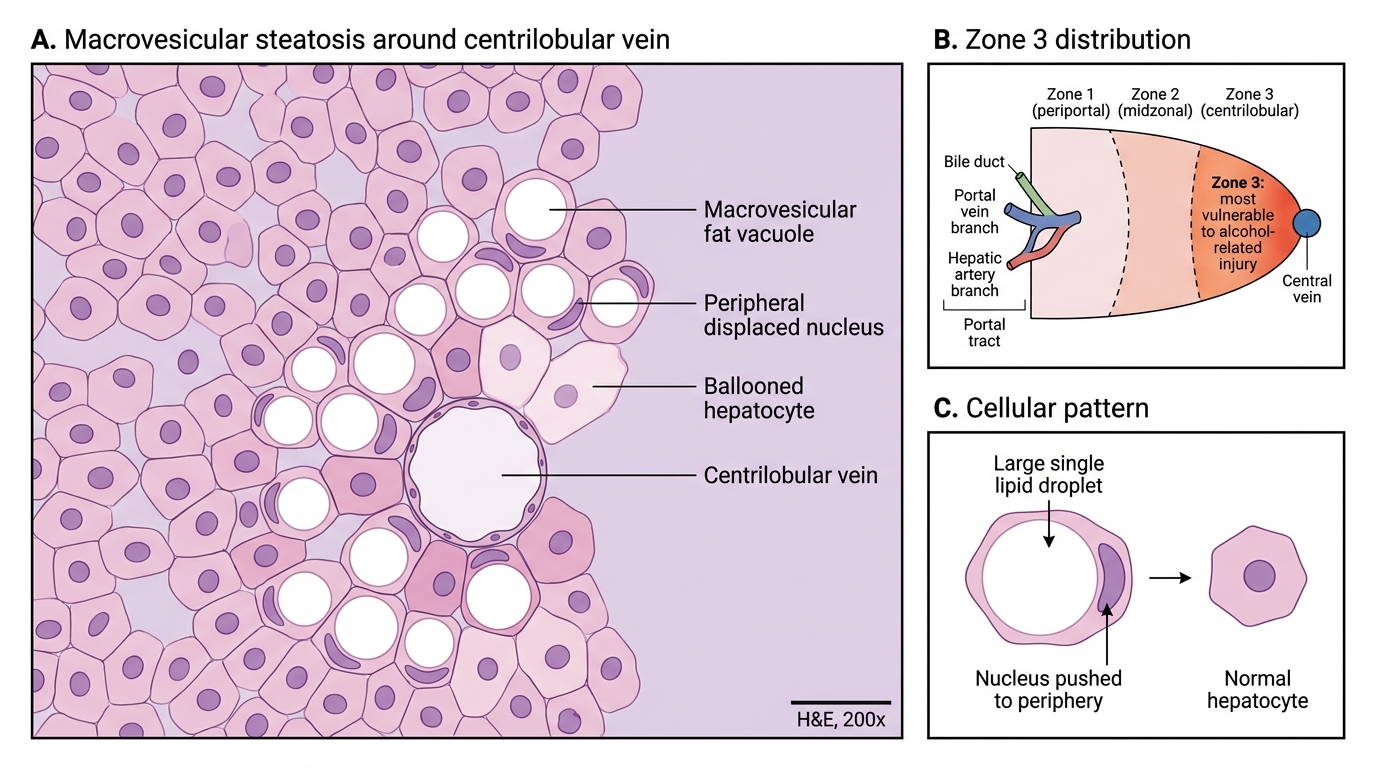
Macrovesicular Steatosis in Zone 3 Liver Injury
Stage 2: Alcoholic Hepatitis
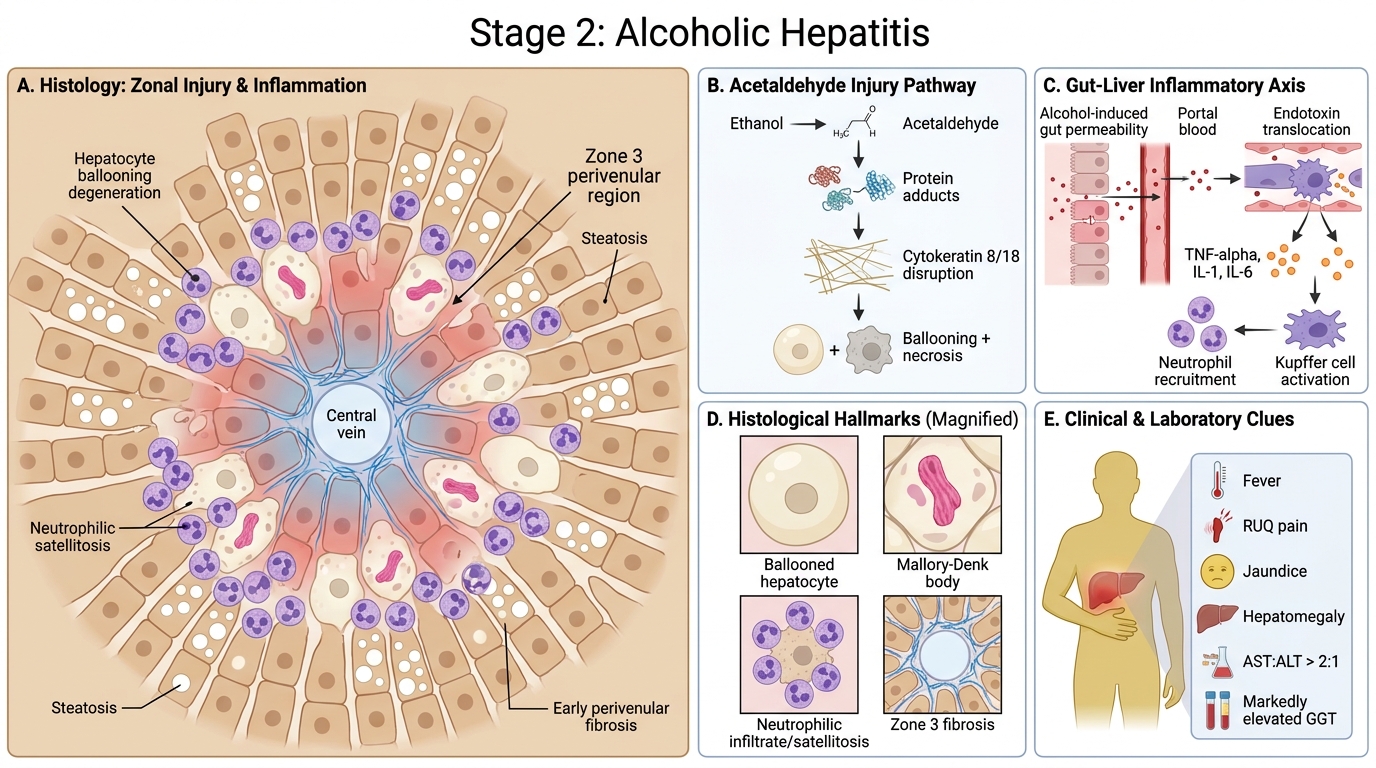
Alcoholic Hepatitis: Pathogenesis, Histology, and Clinical Clues
Alcoholic hepatitis develops with continued alcohol use and represents hepatocyte necrosis superimposed on steatosis.
Pathogenesis: Acetaldehyde forms protein adducts → disrupts cytoskeleton → hepatocyte ballooning and necrosis. Endotoxin translocation from gut (alcohol-induced dysbiosis and increased gut permeability) activates Kupffer cells → release of TNF-α, IL-1, IL-6 → neutrophilic recruitment.
Histological hallmarks — all four must be known:
- Hepatocyte ballooning degeneration — swollen, pale hepatocytes with wispy, cleared cytoplasm (reflects cytoskeletal injury)
- Mallory-Denk bodies (MDB) — irregular, eosinophilic cytoplasmic inclusions composed of misfolded cytokeratin 8/18 intermediate filaments. Pathognomonic of alcoholic hepatitis, though also seen in NASH and Wilson disease. They represent the morphological signature of severe cytoskeletal damage.
- Neutrophilic infiltrate — neutrophils (not lymphocytes) surround and invade damaged hepatocytes (satellitosis) — this neutrophil-dominant pattern distinguishes alcoholic hepatitis from viral hepatitis
- Perivenular (zone 3) fibrosis — early collagen deposition around the central vein (precursor to bridging fibrosis)
Clinical: May present acutely with fever, right upper quadrant pain, jaundice, hepatomegaly. AST:ALT ratio >2:1 is a classic clue (alcohol also inhibits ALT synthesis, a pyridoxal phosphate-dependent enzyme). Serum GGT markedly elevated (alcohol-specific inducer of CYP2E1 pathway).
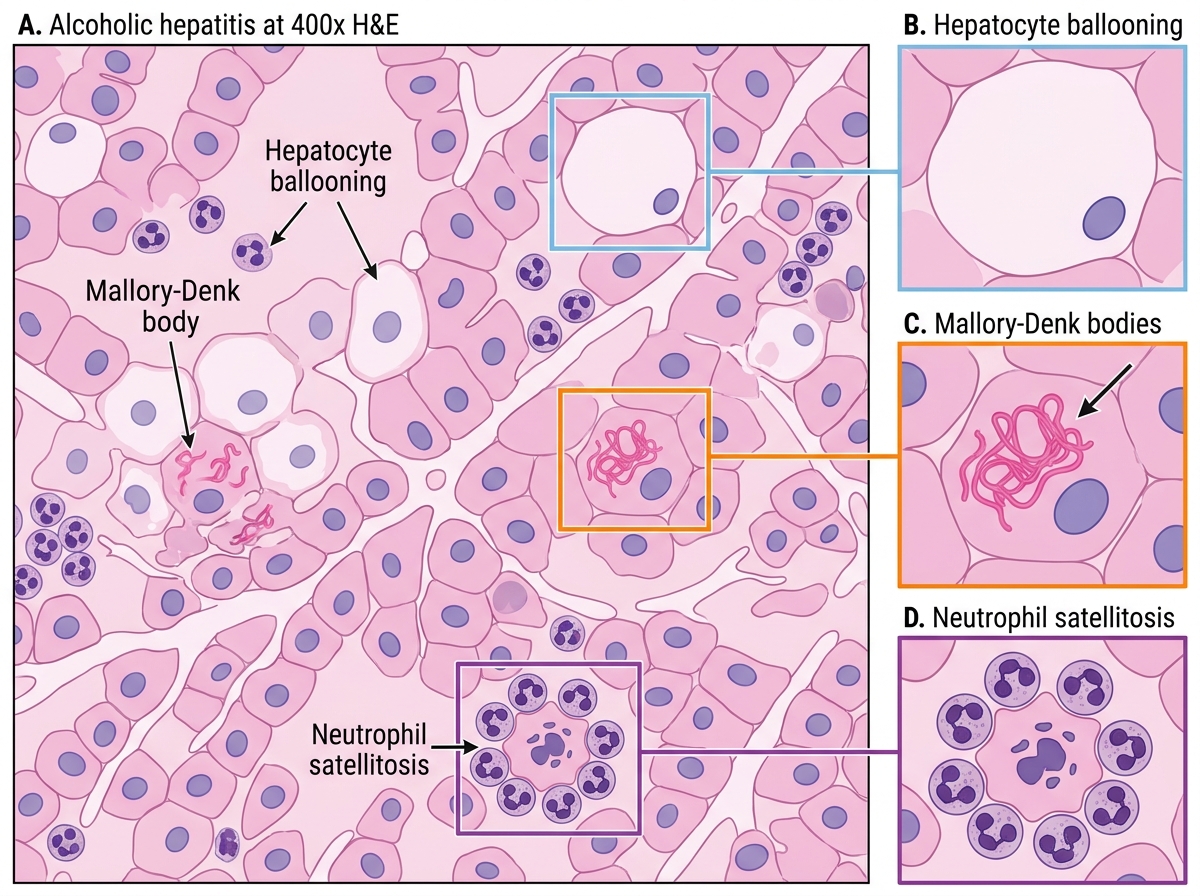
Alcoholic Hepatitis: Key Histological Features
SELF-CHECK
A liver biopsy from a chronic alcohol user shows ballooned hepatocytes with irregular, rope-like eosinophilic cytoplasmic inclusions surrounded by neutrophils. What are these inclusions composed of?
A. Misfolded cytokeratin 8/18 intermediate filaments
B. Aggregated alpha-1-antitrypsin polymers
C. Copper-metallothionein complexes
D. Hepatitis B surface antigen
Reveal Answer
Answer: A. Misfolded cytokeratin 8/18 intermediate filaments
Mallory-Denk bodies are composed of misfolded cytokeratin 8 and 18 intermediate filaments. They are the histological hallmark of alcoholic hepatitis, though they also appear in NASH and Wilson disease. Alpha-1-antitrypsin globules (PAS-positive, diastase-resistant) are seen in alpha-1-antitrypsin deficiency. Copper complexes characterise Wilson disease. HBsAg produces the ground-glass hepatocyte appearance.