Page 7 of 16
PA35.1 | Retinoblastoma & Neuro-Ophthalmic Morphology — SDL Guide
Learning Objectives
- Describe the RB1 tumor suppressor gene, the two-hit hypothesis, and the distinction between hereditary and sporadic retinoblastoma
- Outline the pathogenesis, macroscopic and microscopic morphology of retinoblastoma, including rosette types
- Recognise the clinical presentation, routes of spread, sequelae, and management principles of retinoblastoma
- Identify the key histological features of major CNS/eye tumors in a practical setting: bacterial vs TB meningitis, glioblastoma, meningioma, medulloblastoma, oligodendroglioma, schwannoma, and retinoblastoma
- Contrast small-round-blue-cell tumors (medulloblastoma vs retinoblastoma) and distinguish rosette types (Flexner-Wintersteiner, Homer-Wright, pseudorosette)
INSTRUCTIONS
This is SDL2 of the Nervous System & Eye cluster — the morphology consolidation session. SDL1 covered the theory of CNS tumors and meningitis; today you will build the pattern-recognition library you need for the practical examination. Every lesion in this SDL has a morphological signature. Read actively — as you encounter each description, visualise the slide, then check against the image. By the end, you should be able to look at an unfamiliar glass slide and work through a differential systematically. Retinoblastoma is treated in depth because PA35.1 demands full coverage of etiology through complications; the CNS walk-through consolidates the entire cluster.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch. 28 (Central Nervous System) + Ch. 29 (Eye) (textbook)
- Harsh Mohan, Textbook of Pathology, 8th ed., Ch. 30 (CNS) + Ch. 32 (Eye) (textbook)
- National Retinoblastoma Strategy (Canada/India joint recommendations, 2022 update) — staging, enucleation criteria (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 2-year-old is brought to the paediatric OPD because her mother noticed something strange in a flash photograph: one pupil glows white instead of red. The mother assumed the camera was acting up — until the same white glow appeared in a second photograph three months later. That white reflex, known as leukocoria, is one of medicine's quiet emergencies. The child almost certainly has a malignant tumor growing inside her eye, and the prognosis depends entirely on how quickly the next clinician — perhaps you — recognises what they are seeing. Retinoblastoma is the most common primary intraocular malignancy of childhood. It is also one of the landmark stories in cancer genetics: the tumor that taught us how tumor suppressor genes work and gave us the concept of the 'two-hit hypothesis' — a discovery that changed our understanding of all cancers.
WHY THIS MATTERS
Retinoblastoma is directly mapped to NMC competency PA35.1 and appears consistently in University examinations as a structured question covering genetics, morphology, clinical features, and complications. Beyond the exam, it is clinically urgent: 50% of cases are hereditary, and identifying an index case means screening siblings and offspring can prevent blindness and death. The histology practical will expect you to identify small-round-blue-cell tumors and distinguish rosette types — skills that also apply to medulloblastoma, neuroblastoma, and Ewing sarcoma. The CNS morphology walk-through in this SDL ties together every major lesion from SDL1 and prepares you for the spot-recognition questions that anchor practical marks.
RECALL
From SDL1, you covered the broad categories of CNS tumors: gliomas, meningiomas, nerve sheath tumors, embryonal tumors, and metastases. You also studied bacterial meningitis vs tuberculous meningitis. Recall: glioblastoma is the most malignant astrocytoma (WHO Grade 4); meningioma arises from arachnoid cap cells; medulloblastoma is a cerebellar embryonal tumor. From your Pathology foundation blocks, recall the concept of tumor suppressor genes — genes whose normal function is to brake cell proliferation. Loss-of-function mutations in both alleles (homozygous inactivation) unleash uncontrolled growth. Also recall that retina is embryologically derived from the neuroectoderm — the same layer that forms the brain — which is why retinal tumors share histological features with CNS embryonal tumors.
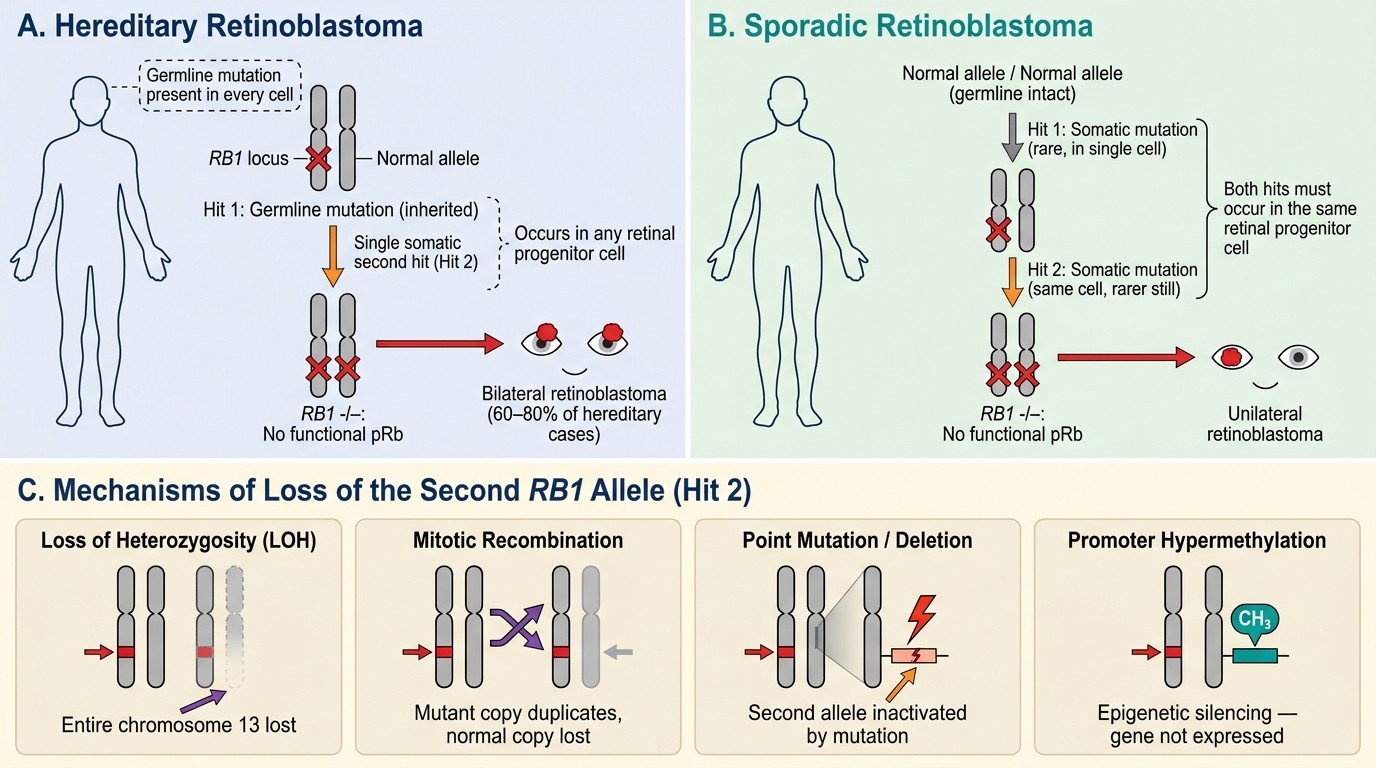
The RB1 Gene and the Two-Hit Hypothesis
Retinoblastoma is caused by biallelic inactivation of the RB1 tumor suppressor gene, located on chromosome 13q14. The RB1 protein (pRb) is a critical regulator of the cell cycle: it restrains progression from G1 into S phase by binding and inactivating the transcription factor E2F. When pRb is inactivated, the cell cannot stop dividing.
Al Knudson proposed the two-hit hypothesis in 1971, based on statistical analysis of retinoblastoma incidence:
- Hit 1 — inactivation of the first RB1 allele (may be germline or somatic)
- Hit 2 — inactivation of the second (remaining normal) allele (always somatic, occurring in a retinal progenitor cell)
Only when both alleles are lost does the cell lose pRb entirely and become malignant.
Knudson Two-Hit Hypothesis: Hereditary vs Sporadic Retinoblastoma and Mechanisms of RB1 Loss
Mechanism of loss of the second allele (Hit 2) — chromosomal mechanisms:
• Loss of heterozygosity (LOH) — the entire chromosome 13 containing the normal allele is lost
• Mitotic recombination — the mutant chromosome duplicates at the expense of the normal copy
• Point mutation or deletion of the second allele
• Promoter hypermethylation (epigenetic silencing)
Downstream effects of RB1 loss:
Without pRb, E2F transcription factors are constitutively active → transcription of genes for DNA synthesis → unrestrained S-phase entry → rapid clonal proliferation of retinal progenitor cells.
Spiral forward (Year 3): The RB1 pathway is disrupted in virtually all human cancers — either through RB1 mutation itself, or through amplification of cyclin D1, loss of p16 (CDKN2A), or CDK4 amplification, all of which converge on hyperphosphorylation and inactivation of pRb.
Hereditary vs Sporadic Retinoblastoma — A Clinically Critical Distinction
Understanding whether a child's retinoblastoma is hereditary or sporadic is essential for prognosis, family screening, and long-term surveillance.
| Feature | Hereditary (Germline) | Sporadic (Somatic) |
|---|---|---|
| Frequency | ~40% of all cases | ~60% of all cases |
| First hit | Germline (present in every cell) | Somatic (in one retinal cell) |
| Second hit required | One somatic event in any retinal cell | Two somatic events in the same cell |
| Laterality | Bilateral (60–80%); may be multifocal | Unilateral, unifocal |
| Age at presentation | Earlier (~12–15 months) | Later (~24–30 months) |
| Family history | Often positive | Usually negative |
| Risk to offspring | 50% (autosomal dominant transmission) | Not hereditary |
| Risk of second cancers | High — especially osteosarcoma, soft-tissue sarcoma, melanoma (radiation-related and non-radiation-related) | Not elevated |
| RB1 testing | Recommended in all first-degree relatives | Germline mutation absent |
Key clinical rule: Any child with bilateral retinoblastoma has hereditary disease by definition. Unilateral disease may be hereditary (5–10%) or sporadic — germline testing is required to distinguish.
Bilateral Retinoblastoma — Fundoscopic Appearance and Endophytic Growth Pattern
The 'new mutation' subset: About 15% of hereditary cases have no family history — the germline mutation arose de novo. These children are the most likely to be missed without high clinical suspicion. Always test for RB1 germline mutation when:
• Bilateral or multifocal disease
• Age < 1 year at diagnosis
• Positive family history of retinoblastoma
Second malignancies in hereditary RB:
Patients who carry a germline RB1 mutation are predisposed to osteosarcoma (most common), soft-tissue sarcomas, melanoma, and brain tumors. The risk is further elevated by radiation therapy (used in prior decades). This is why modern management favours enucleation and chemotherapy over external beam radiation in hereditary cases.
Pathogenesis and Clinical Presentation of Retinoblastoma
Pathogenesis recap:
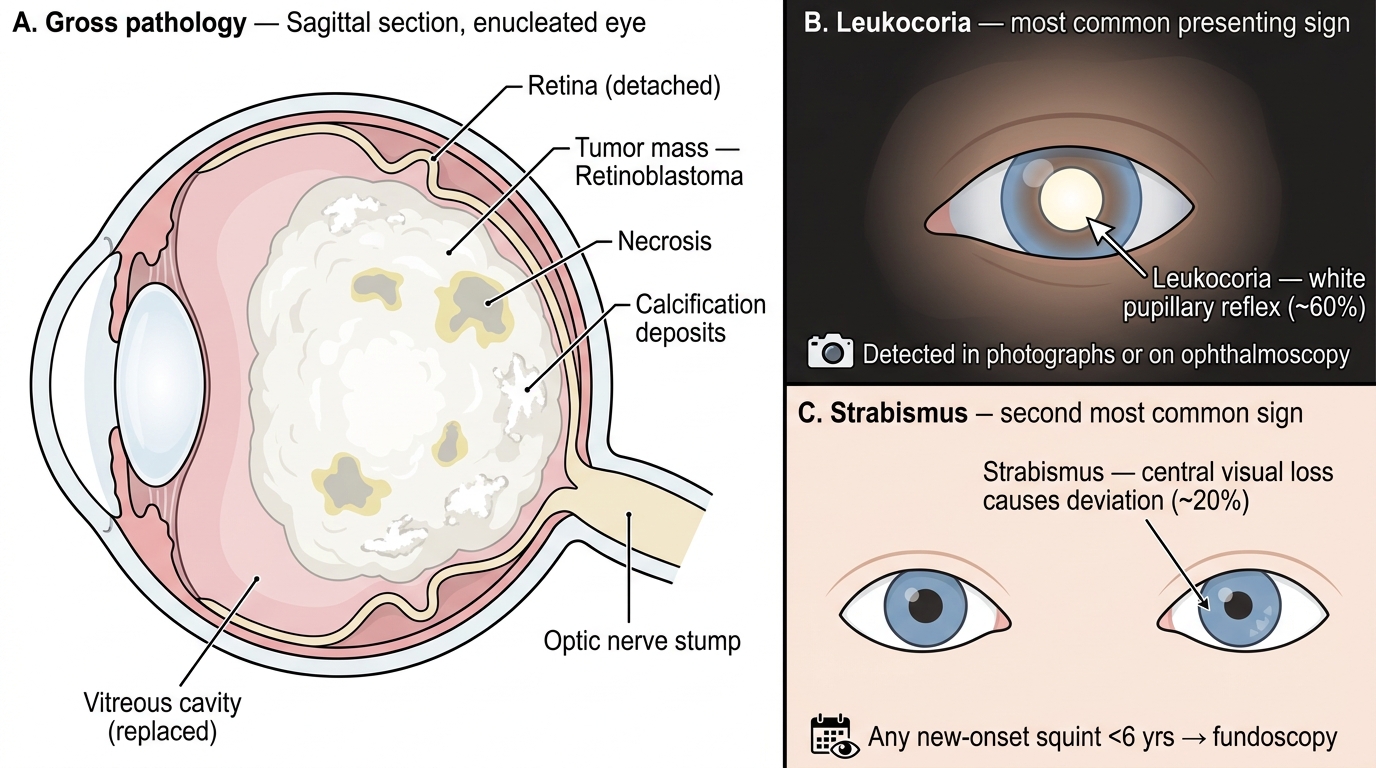
Retinal progenitor cells that lose both RB1 alleles undergo uncontrolled proliferation, forming nests of small, round, poorly differentiated cells. Necrosis is prominent (rapidly growing tumor outstrips its blood supply), and the necrotic debris undergoes dystrophic calcification — a characteristic finding on imaging.
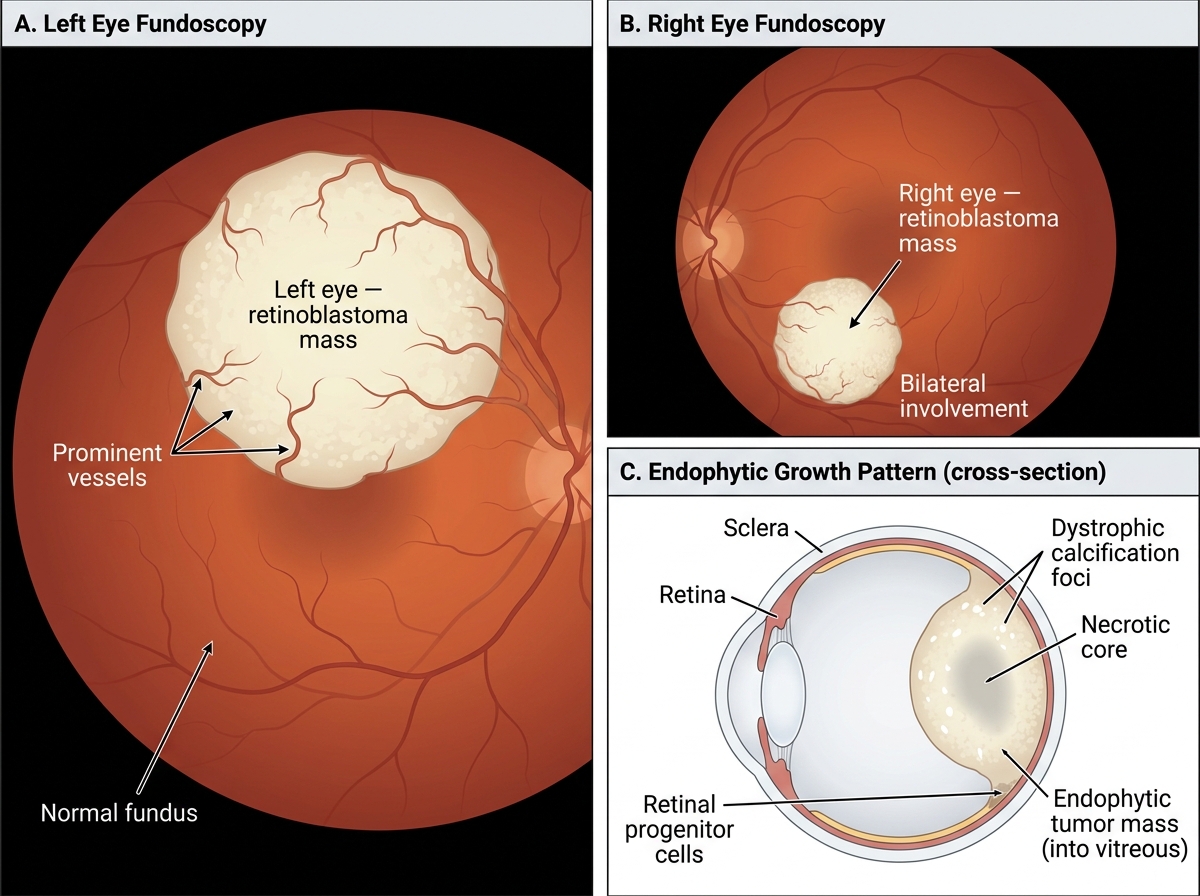
Growth patterns — two macroscopic types:
• Endophytic — grows inward toward the vitreous cavity. White or cream-coloured nodular masses project into the vitreous. Tumor cells may seed the vitreous (vitreous seeding).
• Exophytic — grows outward between the retina and the RPE (retinal pigment epithelium), causing retinal detachment. Subretinal seeding can occur.
• Diffuse infiltrating — rare (~2%); spreads along the retina without forming a discrete mass; diagnosis is most often delayed.
Retinoblastoma — Gross Pathology and Clinical Presentation
Clinical presentation (triad to memorise):
- Leukocoria (white pupillary reflex) — the most common presenting sign (~60% of cases). The tumor replaces the normally transparent vitreous/retina with a white mass, reflecting light as a white glow. Detected by parents in photographs, or by the ophthalmologist with a direct ophthalmoscope in a dark room.
- Strabismus (squint) — second most common (~20%). Central visual loss forces the eye to deviate. Any new-onset strabismus in a child under 6 must prompt fundoscopic examination.
- Others (advanced or unusual presentations): orbital cellulitis-like picture (necrosis-induced inflammation), hyphaema (blood in anterior chamber), heterochromia (different iris colours from neovascularisation), buphthalmos (enlarged eye from raised IOP), or proptosis (orbital extension).
Age: Retinoblastoma is almost exclusively a disease of children under 5 years. Median age at diagnosis: 18 months (bilateral/hereditary) to 24 months (unilateral/sporadic). Cases above age 5 are extremely rare.