Page 15 of 25
PA27.3-4 | Acute & Chronic Renal Failure — SDL Guide
Learning Objectives
- Define acute kidney injury (AKI) and classify it into prerenal, intrinsic, and postrenal categories using clinical and laboratory criteria.
- Distinguish prerenal azotaemia from intrinsic AKI (ATN) using urine indices: FENa, urine osmolality, and urine sodium.
- Describe the four phases of acute tubular necrosis (ATN) and the pathological changes at each phase.
- List the life-threatening complications of AKI: hyperkalaemia, metabolic acidosis, fluid overload, and uraemic syndrome.
- Define chronic kidney disease (CKD), state the KDIGO GFR staging (G1–G5), and identify the five major causes.
- Describe the gross and microscopic pathology of end-stage kidney (contracted, granular, small kidney).
- Explain the pathophysiology of uraemia and enumerate its systemic complications across organ systems.
- Distinguish AKI from CKD using clinical, biochemical, and pathological criteria.
INSTRUCTIONS
Kidney failure sits at the crossroads of medicine — a final pathway for diabetes, hypertension, glomerulonephritis, and obstruction. For Year-2 students who just finished the glomerular and tubular blocks, this module ties those structural lesions to functional collapse. Mastery of the AKI/CKD distinction, the prerenal–ATN urine indices, and the uraemia complication map is essential for your Pathology practical and for clinical reasoning across medicine, surgery, and paediatrics.
References
- Robbins & Kumar Basic Pathology, 11th ed., Ch 14 (textbook)
- Harsh Mohan Textbook of Pathology, 8th ed., Ch 22 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 58-year-old man with type 2 diabetes is admitted in shock after a GI bleed. His serum creatinine on admission is 1.0 mg/dL. Forty-eight hours later it has risen to 4.8 mg/dL and his urine output has fallen to 180 mL/day. The registrar checks his urine sodium — it is 62 mmol/L. The intern asks: 'Is this prerenal failure? Shouldn't his kidneys be holding onto sodium?' The registrar shakes her head: 'No — look at the number. This kidney is no longer capable of doing that.' In the next 21 minutes you will understand exactly why, and what will happen next.
WHY THIS MATTERS
AKI occurs in 10–15% of hospitalised patients and carries an in-hospital mortality of 20–50% in its severe form. CKD affects 850 million people globally — diabetes and hypertension together account for over 50% of cases. These are not rare diseases seen only in nephrology wards. Every clinical posting — medicine, surgery, obstetrics, paediatrics — will bring you face to face with rising creatinine. The concepts in this module (prerenal vs intrinsic distinction, AKI complications, uraemia) appear in the Theory and Practical MBBS examinations and in clinical spotters every year.
RECALL
Before starting, check your readiness:
- From SDL-1 (Normal Kidney): What is the anatomical location of the proximal convoluted tubule, and why is it especially vulnerable to ischaemia?
- From SDL-2 (Glomerular Diseases): What is the nephrotic vs nephritic distinction? Which glomerular diseases progress to CKD?
- From SDL-3 (Tubular & Tubulointerstitial Diseases): How does acute tubular necrosis (ATN) differ histologically from acute interstitial nephritis?
- From Physiology (Year 1): Define GFR. What is the normal range? How is creatinine clearance used to estimate GFR?
- From Biochemistry (Year 1): What buffering system does the kidney use to maintain pH? What happens to bicarbonate in renal failure?
If any of these feel uncertain, spend 3–4 minutes reviewing before proceeding — this module builds directly on that foundation.
Acute Kidney Injury — Definition and Classification
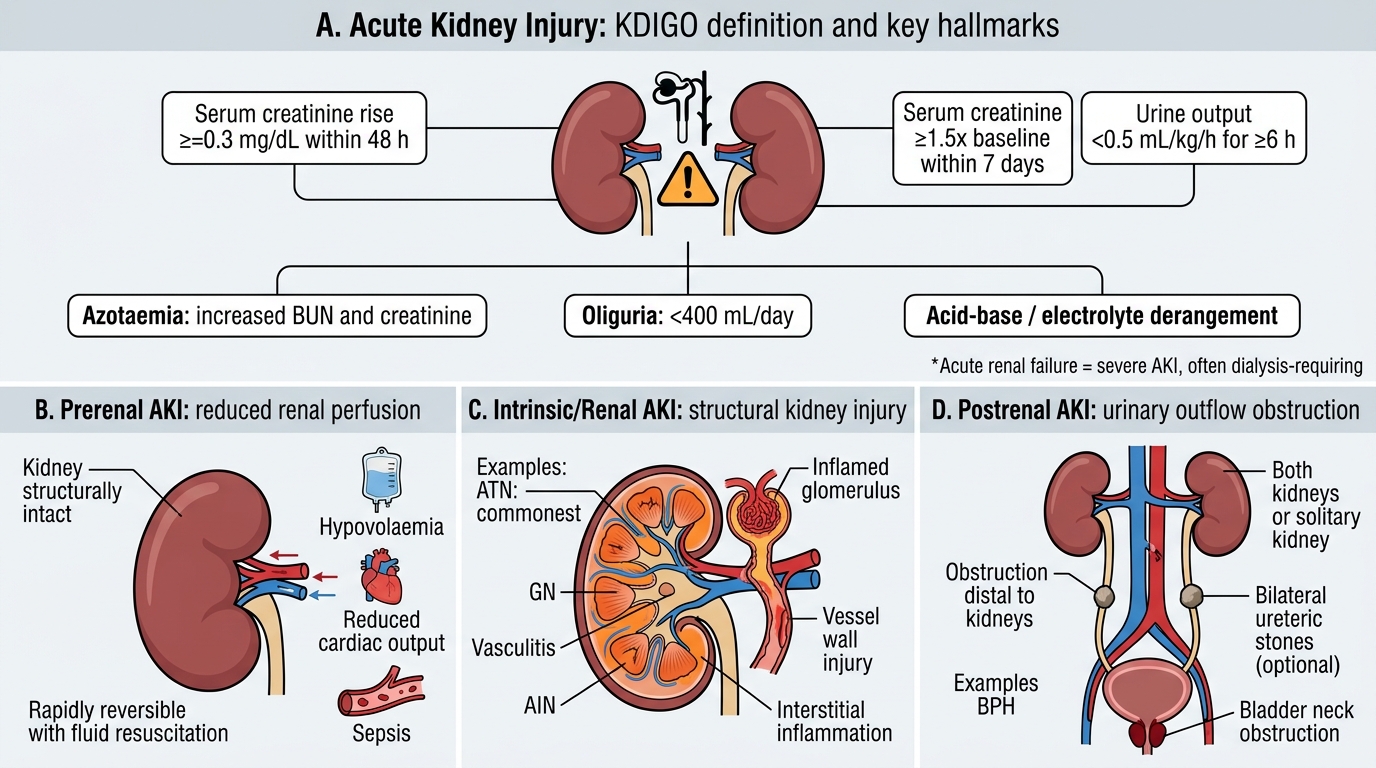
Acute Kidney Injury: Definition and Anatomical Classification
Acute kidney injury (AKI) is defined by the KDIGO criteria as any of the following: (1) rise in serum creatinine ≥0.3 mg/dL within 48 hours; (2) rise in serum creatinine ≥1.5× baseline within 7 days; or (3) urine output <0.5 mL/kg/h for ≥6 hours. The older term 'acute renal failure' is reserved for the severe end (requiring dialysis). The hallmarks are azotaemia (↑BUN and creatinine), oliguria (<400 mL/day), and acid-base/electrolyte derangement.
AKI is classified by the anatomical site of insult — a framework that drives management:
| Class | Mechanism | Key Examples |
|---|---|---|
| Prerenal | ↓Renal perfusion; kidneys structurally intact | Hypovolaemia, ↓CO (heart failure), sepsis |
| Intrinsic/Renal | Structural damage to glomeruli, tubules, vessels, or interstitium | ATN (commonest), GN, vasculitis, AIN |
| Postrenal | Obstruction to urine outflow (both kidneys or solitary) | BPH, bilateral ureteric stones, bladder neck obstruction |
Recognising the class is urgent because prerenal AKI is rapidly reversible with fluid resuscitation, while established ATN is not.
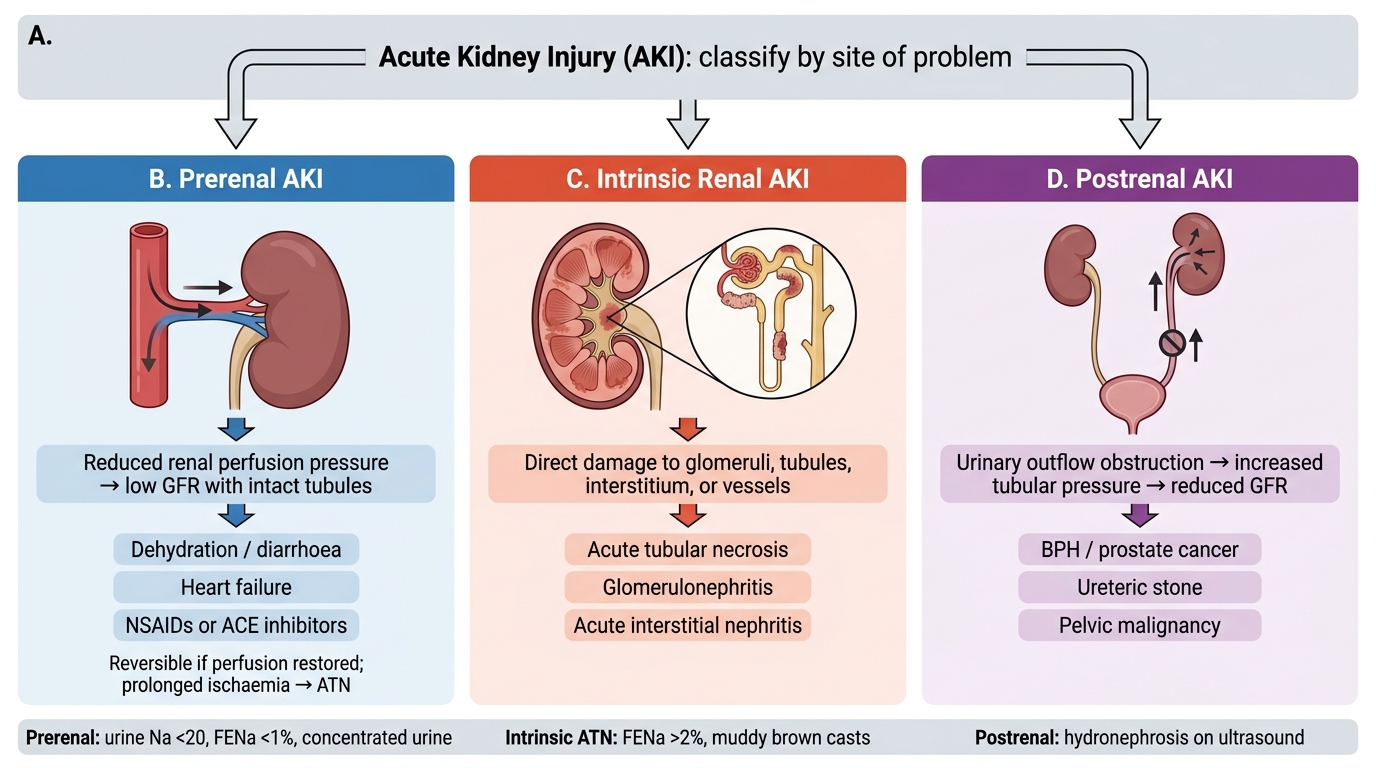
Classification of Acute Kidney Injury
Prerenal AKI — Pathophysiology and Reversibility
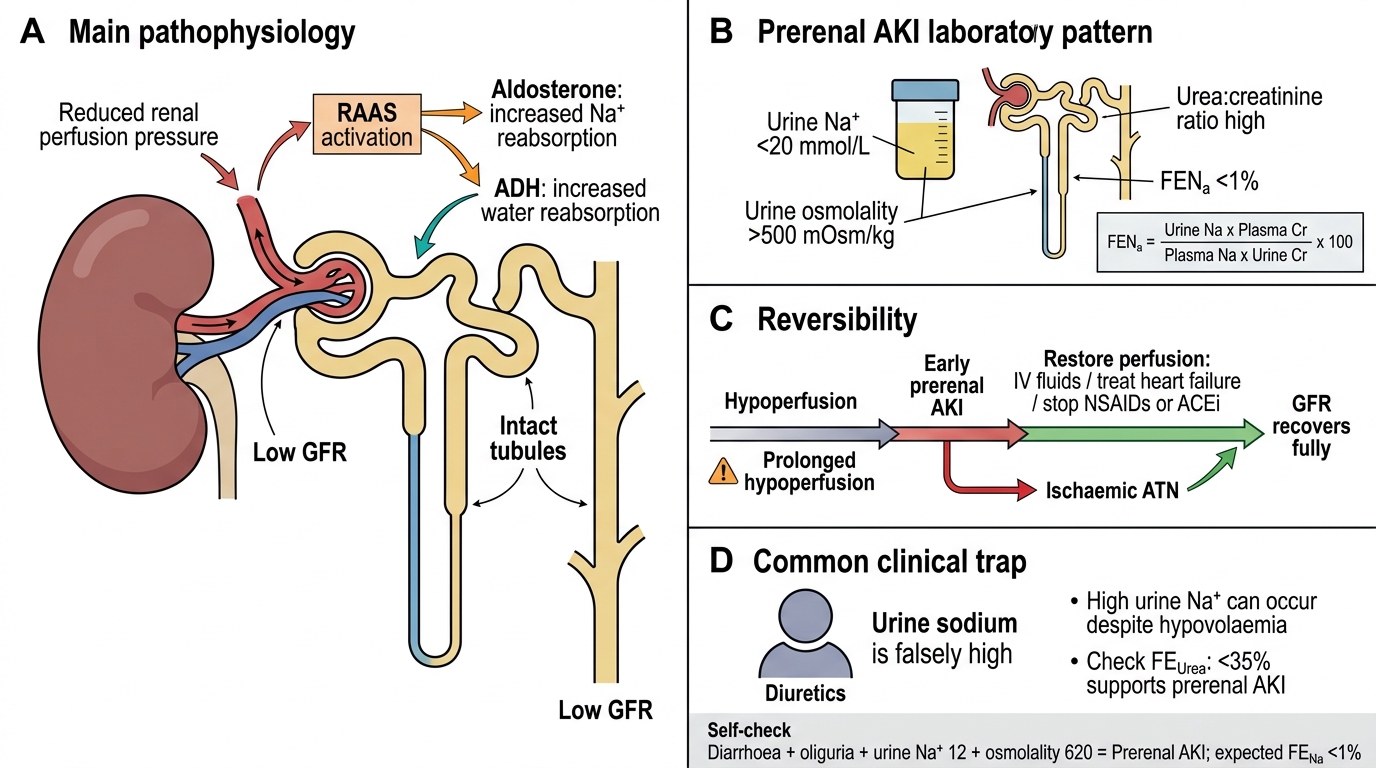
Prerenal AKI: Intact Tubules, Sodium Retention, and Reversibility
In prerenal AKI, reduced perfusion pressure activates the renin-angiotensin-aldosterone system (RAAS) and ADH. The tubules are structurally intact and respond normally:
- Aldosterone → ↑Na reabsorption → urine Na low (<20 mmol/L)
- ADH → ↑water reabsorption → urine osmolality high (>500 mOsm/kg)
- Urea is reabsorbed passively with water → urea:creatinine ratio high (>60:1 using mmol, or >20:1 using mg)
The fractional excretion of sodium (FENa) quantifies sodium handling:
FENa = (Urine Na × Plasma Cr) / (Plasma Na × Urine Cr) × 100
In prerenal AKI: FENa <1% (tubules avidly reabsorbing sodium).
Reversibility: If perfusion is restored quickly (IV fluids, treating heart failure, stopping NSAIDs/ACEi), GFR recovers fully. Prolonged prerenal ischaemia → progression to ischaemic ATN.
Clinical trap: Patients on diuretics may have high urine Na despite true hypovolaemia — check fractional excretion of urea (FEUrea) instead (<35% = prerenal).
SELF-CHECK
A 70-year-old woman presents with diarrhoea for 3 days and oliguria. Serum creatinine is 2.8 mg/dL (baseline 0.9). Urine sodium is 12 mmol/L, urine osmolality is 620 mOsm/kg. What is the most likely diagnosis and expected FENa?
A. Acute tubular necrosis; FENa >2%
B. Prerenal AKI; FENa <1%
C. Acute interstitial nephritis; FENa 1–2%
D. Postrenal AKI; FENa variable
Reveal Answer
Answer: B. Prerenal AKI; FENa <1%
Low urine Na (12 mmol/L), high urine osmolality (620 mOsm/kg), and a clear precipitant (volume depletion from diarrhoea) indicate prerenal AKI — structurally intact tubules avidly reabsorbing sodium under RAAS/ADH drive. FENa <1% confirms this. ATN is characterised by FENa >2% and urine Na >40 mmol/L because damaged tubules can no longer reabsorb sodium.
Intrinsic AKI — ATN: Causes and Pathology
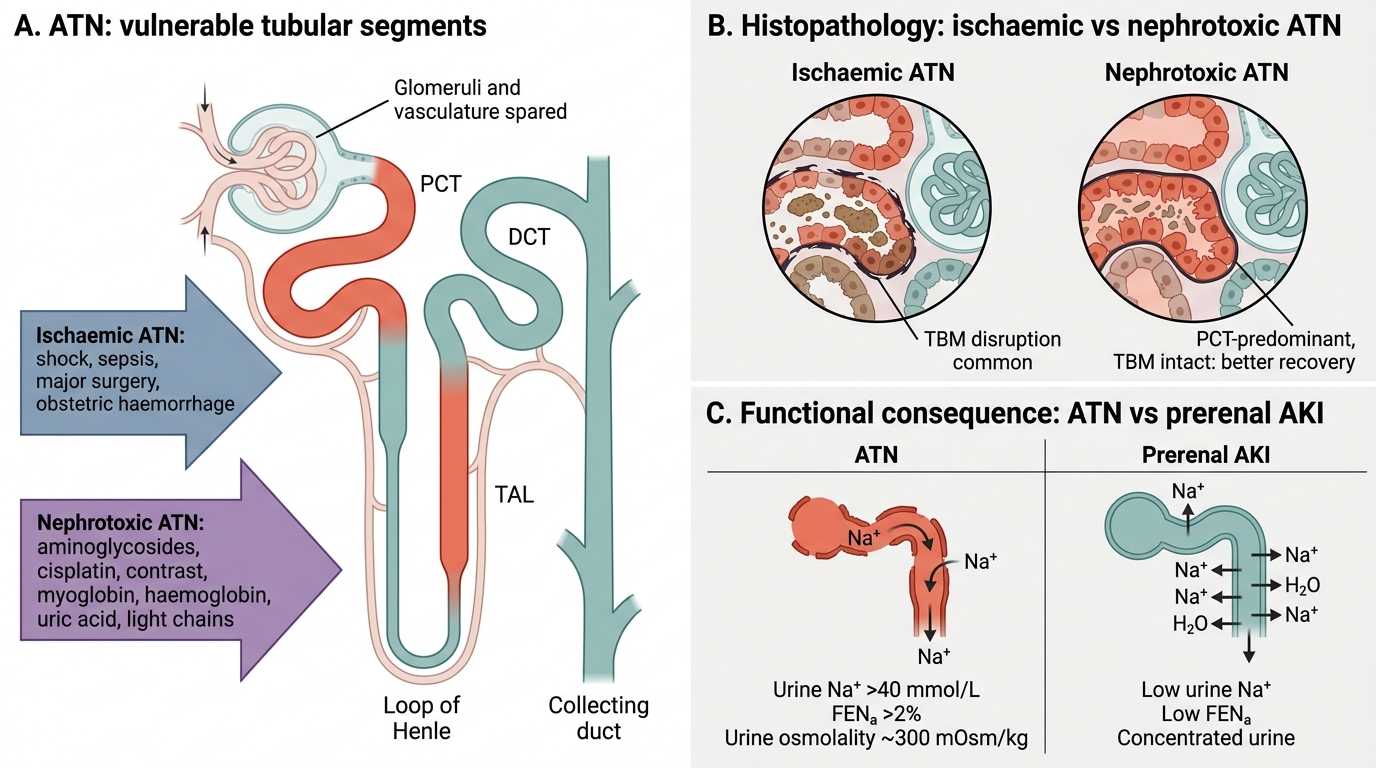
Acute Tubular Necrosis: Causes, Pathology, and Urine Findings
Acute tubular necrosis (ATN) is the commonest cause of intrinsic AKI (accounting for ~75% of all AKI). There are two major pathways:
1. Ischaemic ATN — sustained prerenal ischaemia (shock, sepsis, major surgery, obstetric haemorrhage). The proximal tubule (PCT) and the thick ascending limb of Henle (TAL) — both high-metabolic-rate, oxygen-dependent — are most vulnerable.
2. Nephrotoxic ATN — direct tubular toxicity:
- Exogenous: aminoglycosides, cisplatin, contrast agents, myoglobin (rhabdomyolysis), haemoglobin (haemolysis)
- Endogenous: uric acid (tumour lysis syndrome), light chains (myeloma cast nephropathy)
Histopathology of ATN:
- Patchy tubular epithelial necrosis and sloughing into the lumen
- Tubular basement membrane disruption (more in ischaemic ATN)
- Tubular casts — granular, pigmented (in rhabdomyolysis: myoglobin casts; in haemolysis: haemoglobin casts)
- Note: glomeruli and vasculature are spared — this is a pure tubular disease
- In nephrotoxic ATN: necrosis is typically confined to the PCT (proximal tubule most exposed to filtered toxin); basement membrane intact → better recovery potential
In contrast to prerenal: ATN tubules cannot reabsorb sodium → urine Na >40 mmol/L, FENa >2%, urine osmolality low (~300 mOsm/kg, isosthenuric).