Page 6 of 25
PA27.5-6 | Glomerular Diseases — SDL Guide
Learning Objectives
- Classify glomerular diseases into nephrotic and nephritic syndromes with their defining features
- Explain the four major mechanisms of glomerular injury with examples of each
- Correlate LM, IF, and EM findings for each major glomerular disease
- Describe the pathology, clinical features, and complement profile of post-streptococcal GN and RPGN
- Explain IgA nephropathy (Berger disease) in depth — pathogenesis, synpharyngitic haematuria, pathology, progression, and Henoch-Schönlein link
INSTRUCTIONS
Glomerular diseases are the commonest cause of chronic kidney disease worldwide, and Year-2 Pathology examinations consistently test the ability to distinguish nephrotic from nephritic syndrome, interpret the LM/IF/EM triad, and reason through complement levels and deposit patterns. This module builds from mechanisms of injury (the 'why') to disease-specific pathology (the 'what'), ending with IgA nephropathy — the single commonest GN globally. Master this module and you will hold a robust diagnostic framework for the entire glomerular disease spectrum.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th Ed, Ch 20 (textbook)
- Harsh Mohan Textbook of Pathology, 7th Ed, Ch 22 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 6-year-old boy presents with swollen eyelids each morning, frothy urine, and no haematuria. A 35-year-old man presents with cola-coloured urine two days after a sore throat. Both patients have kidney disease — but their kidneys look completely different under the microscope, and they need completely different treatments. The first child will respond to steroids and fully recover. The second patient needs urgent investigation to prevent renal failure. The distinction between these two worlds — nephrotic syndrome and nephritic syndrome — is the foundation of glomerular pathology.
WHY THIS MATTERS
Glomerular diseases are among the top three causes of end-stage renal disease globally. In India, IgA nephropathy and post-infectious GN are particularly prevalent, making their recognition a core clinical competency. The PA27.5 and PA27.6 competencies you complete today map directly to the NMC CBME exit-level requirement that every graduating doctor can interpret a renal biopsy report and correlate it with the clinical syndrome.
RECALL
Before proceeding, recall from your Year-1 studies:
• The glomerular filtration barrier has three layers: fenestrated endothelium, glomerular basement membrane (GBM), and podocytes (visceral epithelial cells) with their foot processes.
• Normal urine protein < 150 mg/day; heavy proteinuria (> 3.5 g/day) signals barrier disruption.
• The complement system (C3, C4, CH50) is consumed during immune-complex activation — a falling complement level is a clue to immune-mediated disease.
• Review the basic anatomy of Bowman's capsule, the mesangium, and the capillary loops — these spatial relationships explain why deposits in different compartments produce different patterns.
Mechanisms of Glomerular Injury
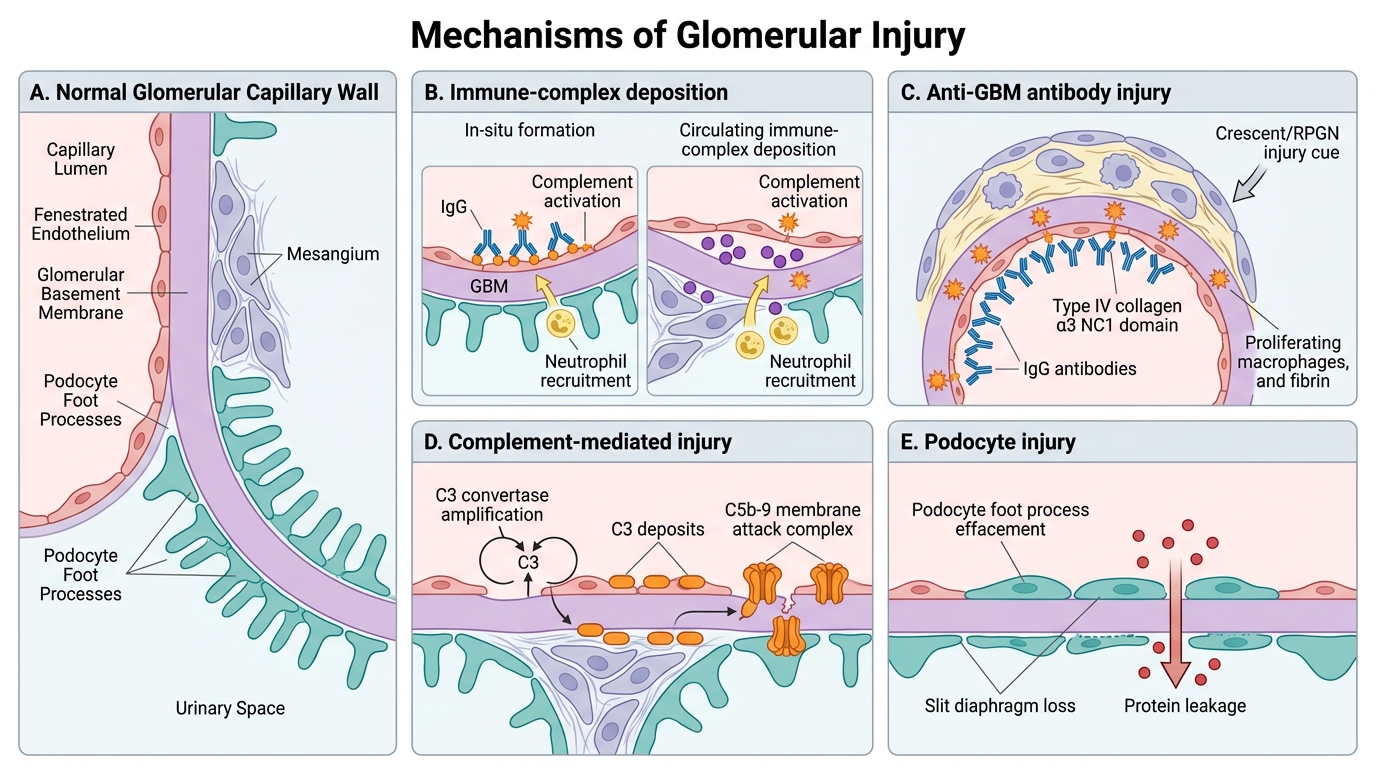
Mechanisms of Glomerular Injury
The glomerulus is injured by four principal mechanisms. Understanding these mechanisms predicts which diseases activate complement, which spare it, and which require biopsy for diagnosis.
1. Immune-complex deposition — two routes
In-situ formation: Circulating antibodies bind to antigens already planted in the glomerulus (e.g., PLA2R antigen in membranous nephropathy, streptokinase antigen in post-infectious GN). The complex forms locally, activating complement directly on the GBM or mesangium.
Circulating immune-complex deposition: Preformed antigen–antibody complexes deposit from the bloodstream in the mesangium or subendothelial space (e.g., lupus nephritis, IgA nephropathy). Complement is activated and mediates injury via the membrane attack complex (C5b-9) and neutrophil recruitment.
2. Anti-GBM antibodies
Autoantibodies target type IV collagen, specifically the NC1 domain of the α3 chain. These antibodies bind linearly along the entire GBM. The result is a linear IF pattern — the pathognomonic fingerprint of Goodpasture syndrome and Type I RPGN. Complement is activated intensely, causing severe, rapid injury.
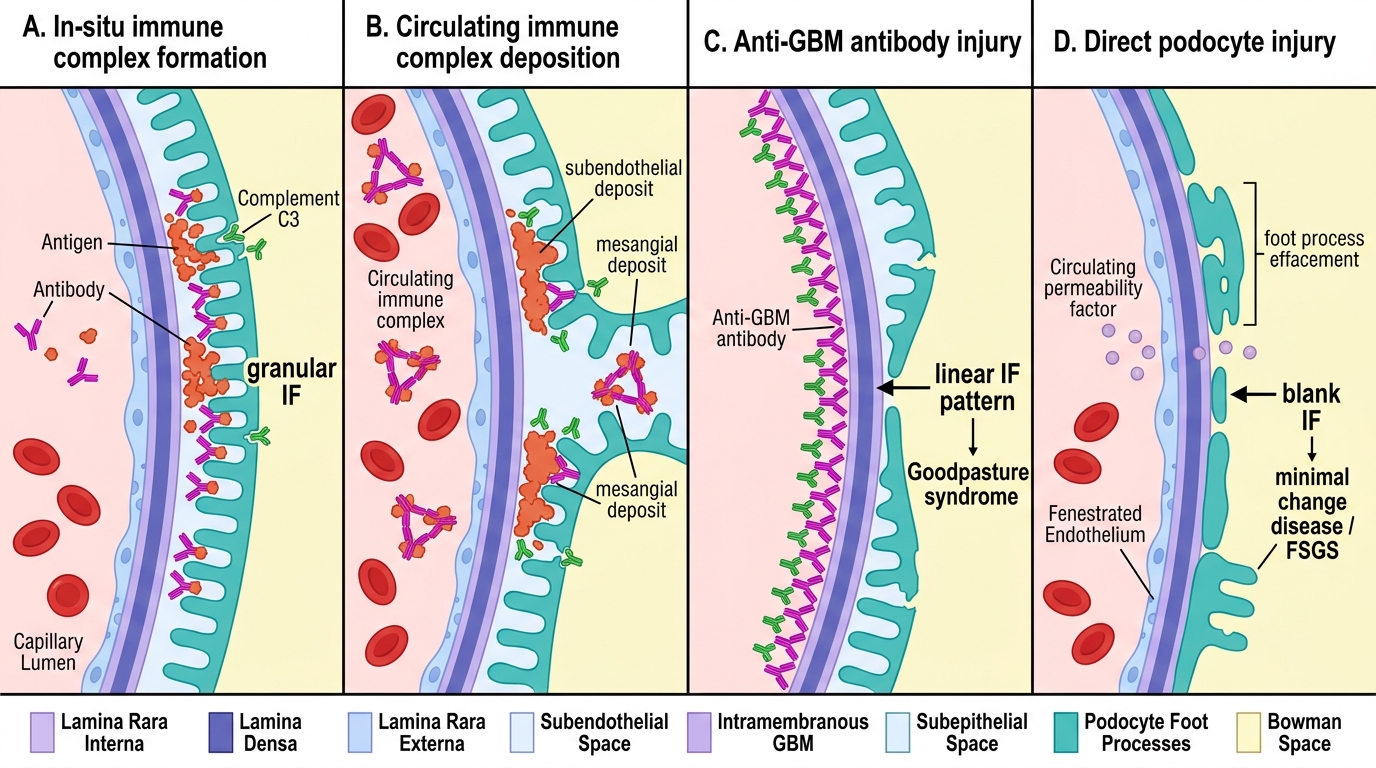
Major Mechanisms of Glomerular Injury
3. Complement-mediated injury (alternative pathway)
Mutations in complement regulatory proteins (factor H, factor I) allow uncontrolled C3 activation at the GBM. This underlies dense deposit disease (a form of MPGN). C3 deposits are seen without Ig on IF.
4. Direct podocyte injury
Circulating permeability factors (e.g., suPAR in FSGS, unknown factor in MCD) directly disrupt podocyte foot processes. There is no immune-complex deposition and no complement activation. This explains why minimal change disease (MCD) has a normal LM and a blank IF — yet massive proteinuria on clinical presentation.
| Mechanism | IF pattern | Complement | Example |
|---|---|---|---|
| In-situ IC | Granular | ↓ | Membranous NP, PSGN |
| Circulating IC | Granular | ↓ (variable) | Lupus, IgA NP |
| Anti-GBM | Linear | ↓ | Goodpasture |
| Podocyte injury | Negative | Normal | MCD, FSGS |
SELF-CHECK
A renal biopsy shows linear IgG deposits along the GBM on immunofluorescence. Which mechanism best explains this pattern?
A. Circulating immune-complex deposition in the mesangium
B. In-situ immune-complex formation at the subepithelial space
C. Autoantibodies directed against type IV collagen in the GBM
D. Direct podocyte injury by a circulating permeability factor
Reveal Answer
Answer: C. Autoantibodies directed against type IV collagen in the GBM
Linear IgG staining is the hallmark of anti-GBM antibody disease (Type I RPGN / Goodpasture syndrome). The antibody binds uniformly along the entire length of the GBM, producing the continuous linear rather than granular or segmental pattern seen with immune-complex deposition.
Nephrotic Syndrome: Overview and Pathophysiology
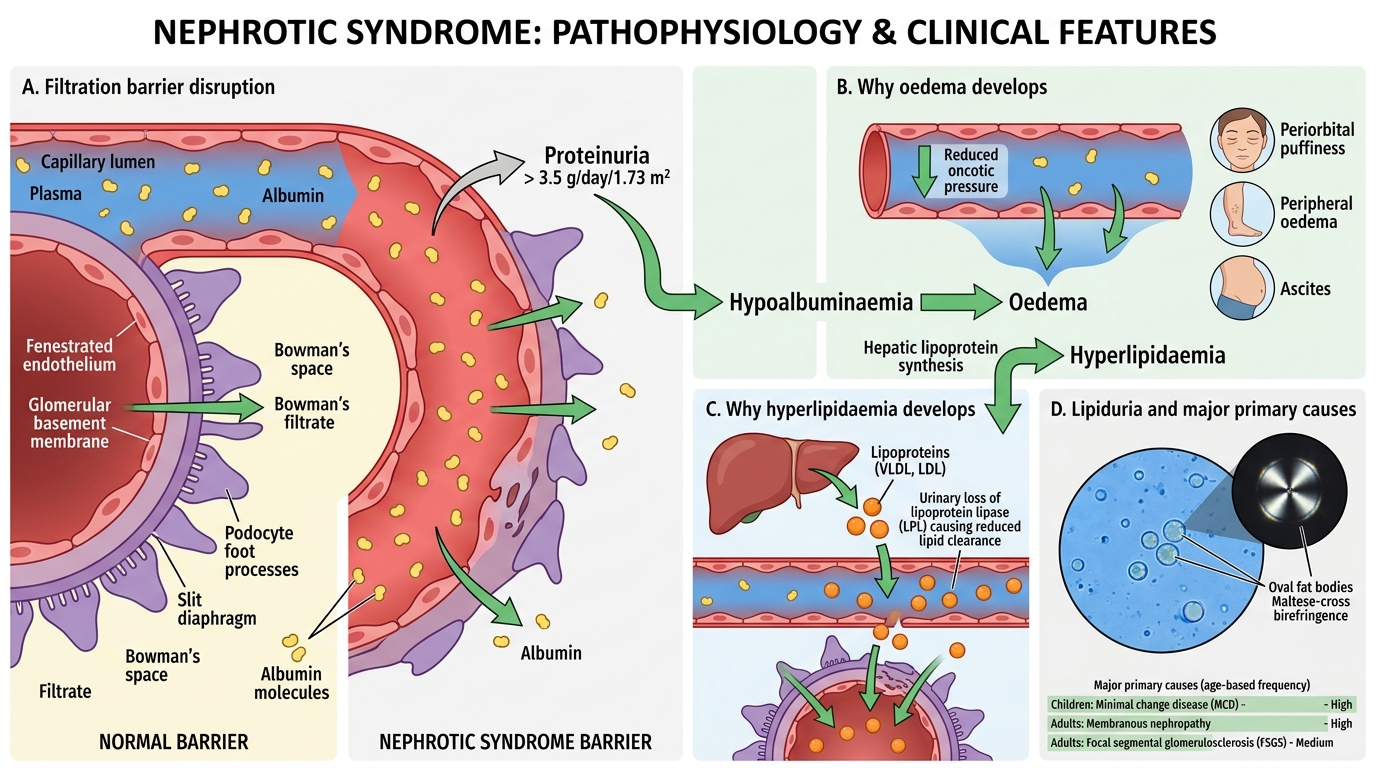
Nephrotic Syndrome: Pathophysiology Overview
Nephrotic syndrome is defined by: proteinuria > 3.5 g/day/1.73 m², hypoalbuminaemia, generalised oedema, hyperlipidaemia, and lipiduria. The unifying defect is disruption of the filtration barrier, allowing albumin and other plasma proteins to pour into the filtrate.
Why oedema? Hypoalbuminaemia reduces oncotic pressure → fluid shifts to interstitium → ascites, peripheral oedema, periorbital puffiness (most prominent in the morning when albumin redistribution is maximal after recumbency).
Why hyperlipidaemia? Reduced oncotic pressure triggers hepatic lipoprotein synthesis (compensatory). Urinary loss of lipoprotein lipase leads to impaired clearance.
Why lipiduria? Lipoproteins leak through the damaged filter and appear as oval fat bodies and Maltese-cross birefringence in urine under polarised light.
The four major primary causes in decreasing age-specific frequency:
- Children (< 10 yr): Minimal change disease (MCD) ~90%
- Adults: Membranous nephropathy (most common primary, 35-40%)
- Focal segmental glomerulosclerosis (FSGS)
- Membranoproliferative GN (MPGN)
Minimal Change Disease (MCD)
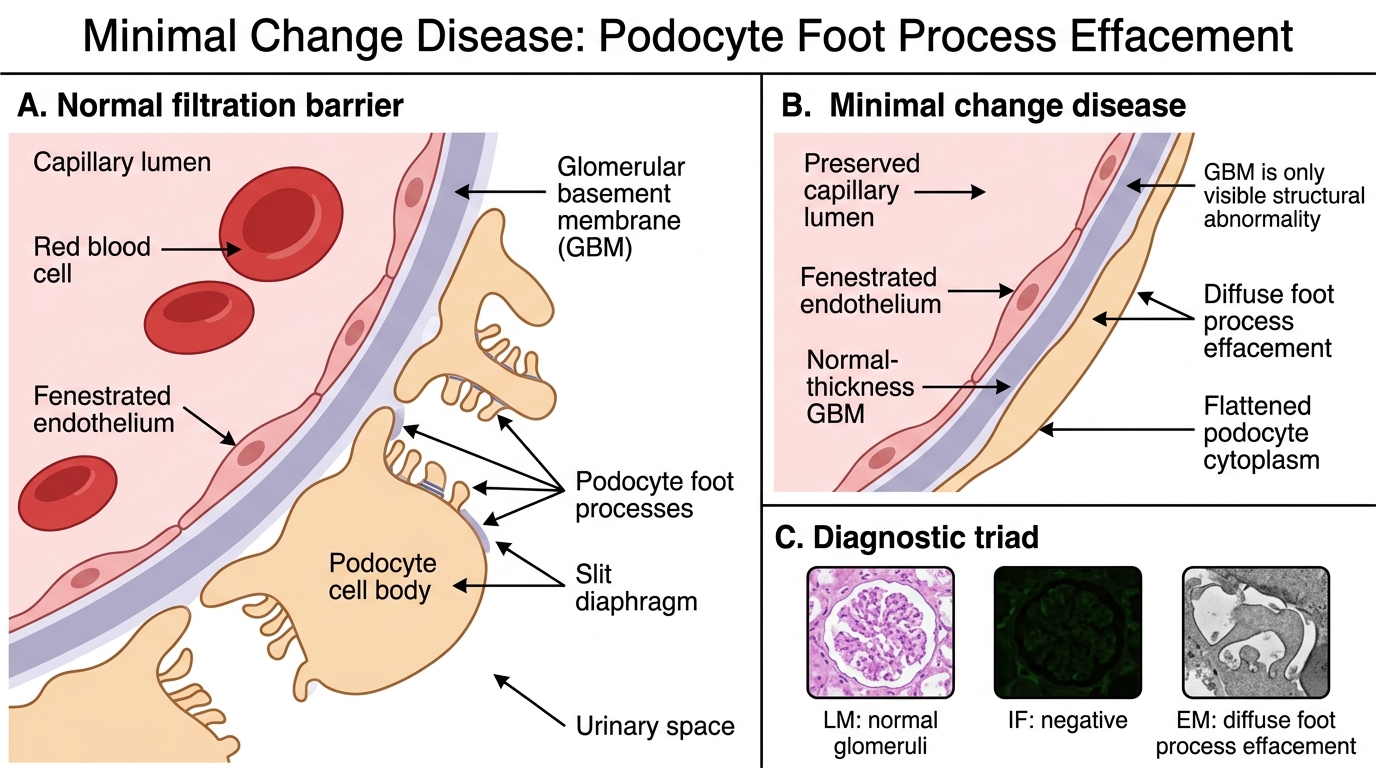
Minimal Change Disease: Foot Process Effacement
Minimal change disease is the paradigm of podocyte injury without structural change visible by LM or IF.
Who gets it? Children aged 1-8 years; peak at ~3 years. Accounts for ~90% of childhood nephrotic syndrome. Rare in adults but associated with Hodgkin lymphoma (lymphokine-mediated podocyte injury) and NSAIDs.
Pathology — the diagnostic triad:
• LM: Completely normal glomeruli — no hypercellularity, no GBM thickening. This is a trap: the child has massive proteinuria yet the biopsy looks normal.
• IF: Negative — no immunoglobulin or complement deposition.
• EM: Diffuse effacement of podocyte foot processes across the entire glomerular surface. This is the ONLY structural abnormality. Foot process effacement = the diagnostic lesion.
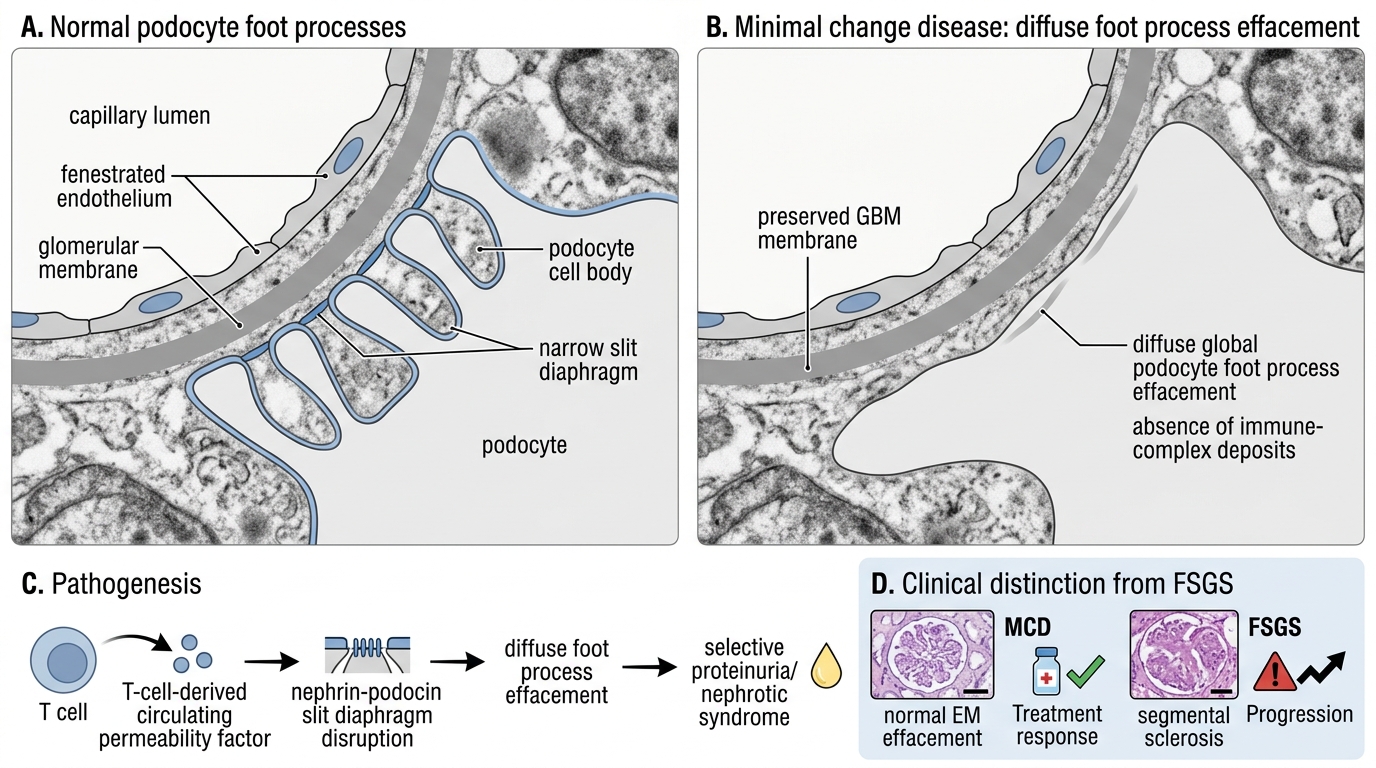
Minimal Change Disease: Podocyte Foot Process Effacement
Pathogenesis: A T-cell–derived circulating permeability factor (likely suPAR or a related lymphokine) disrupts the nephrin–podocin slit diaphragm, causing diffuse foot process effacement without immune-complex formation.
Clinical course: Exquisitely steroid-responsive — 95% of children enter complete remission within 8 weeks. Relapses occur (~50%) but prognosis is excellent; does NOT progress to CKD. This steroid responsiveness is diagnostically useful: if a child with nephrotic syndrome does not respond to steroids, suspect FSGS.
Key distinguishing feature vs FSGS: MCD = diffuse, global foot process effacement, normal LM, excellent prognosis. FSGS = segmental sclerosis on LM, poor prognosis.