Page 11 of 25
PA27.7 | Glomerular Manifestations of Systemic Disease — SDL Guide
Learning Objectives
- Enumerate the major systemic diseases that cause glomerular injury and classify their mechanisms of damage
- Describe the pathogenesis, morphology, and clinical stages of diabetic nephropathy including Kimmelstiel-Wilson nodular glomerulosclerosis
- Apply the ISN/RPS classification of lupus nephritis (classes I–VI) and identify the hallmark lesions of class IV diffuse proliferative lupus nephritis
- Recognise the renal manifestations of amyloidosis, hypertensive nephrosclerosis (benign and malignant), and other systemic conditions
- Interpret the clinical syndrome — proteinuria, haematuria, renal function decline — within the context of an underlying systemic disease
INSTRUCTIONS
Systemic diseases are the single largest contributor to end-stage renal disease (ESRD) worldwide. Unlike primary glomerulonephritides, these conditions arrive with a diagnosis already in hand — diabetes, lupus, hypertension — yet renal involvement frequently determines prognosis and survival. Understanding how a dysregulated systemic milieu translates into glomerular injury is the bridge between your Medicine and Pathology teaching, and a direct requirement of PA27.7. This module will take you through the major offenders, their mechanisms, their microscopic signatures, and the clinical syndromes they produce.
References
- Robbins & Kumar: Basic Pathology, 11th ed., Ch 14 (The Kidney) (textbook)
- Harsh Mohan: Textbook of Pathology, 8th ed., Ch 23 (textbook)
- Chandrasoma & Taylor: Concise Pathology, 3rd ed., Ch 22 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 52-year-old woman with a 15-year history of type 2 diabetes mellitus presents with progressively worsening leg oedema. Urine dipstick shows 3+ protein. Her serum creatinine is 3.2 mg/dL, up from a baseline of 1.1 mg/dL three years ago. Her nephrologist tells her she is in stage 4 chronic kidney disease and may need dialysis within the year. No kidney biopsy is done — the clinical picture is diagnostic. Why? And what would a biopsy show if one were performed? This module answers that question, and extends it to lupus, amyloid, hypertension, and a clutch of other systemic destroyers of glomeruli.
WHY THIS MATTERS
Diabetic nephropathy is the leading cause of ESRD in India and worldwide — accounting for over 40% of new dialysis patients. Lupus nephritis determines survival in SLE. Malignant hypertension can produce acute kidney injury that is histologically distinctive. For a clinician, recognising renal involvement in systemic disease early — through a urinalysis, a serum creatinine, or a urine albumin-to-creatinine ratio — is the difference between slowing progression and watching irreversible fibrosis accumulate. Pathology underpins that recognition: you cannot interpret clinical findings without knowing what the glomerulus looks like under each insult.
RECALL
Before you begin, recall from earlier modules:
- Glomerular filtration barrier — endothelium (fenestrated), glomerular basement membrane (GBM), podocytes (foot processes, slit diaphragm). Each layer has a role in restricting protein loss.
- Mesangium — mesangial cells + matrix; supports the capillary loops; expands in response to injury and immune deposits.
- Nephrotic vs nephritic syndrome — nephrotic: heavy proteinuria (>3.5 g/day), hypoalbuminaemia, oedema, hyperlipidaemia; nephritic: haematuria, mild-to-moderate proteinuria, hypertension, oliguria, RBC casts.
- Immune-complex deposition — complement activation, mesangial and subendothelial/subepithelial deposits; basis of lupus and IgA nephropathy.
- From Year-1 Physiology: renin-angiotensin-aldosterone axis, autoregulation of glomerular pressure — both hijacked in diabetes and hypertension.
Diabetic Nephropathy — Pathogenesis
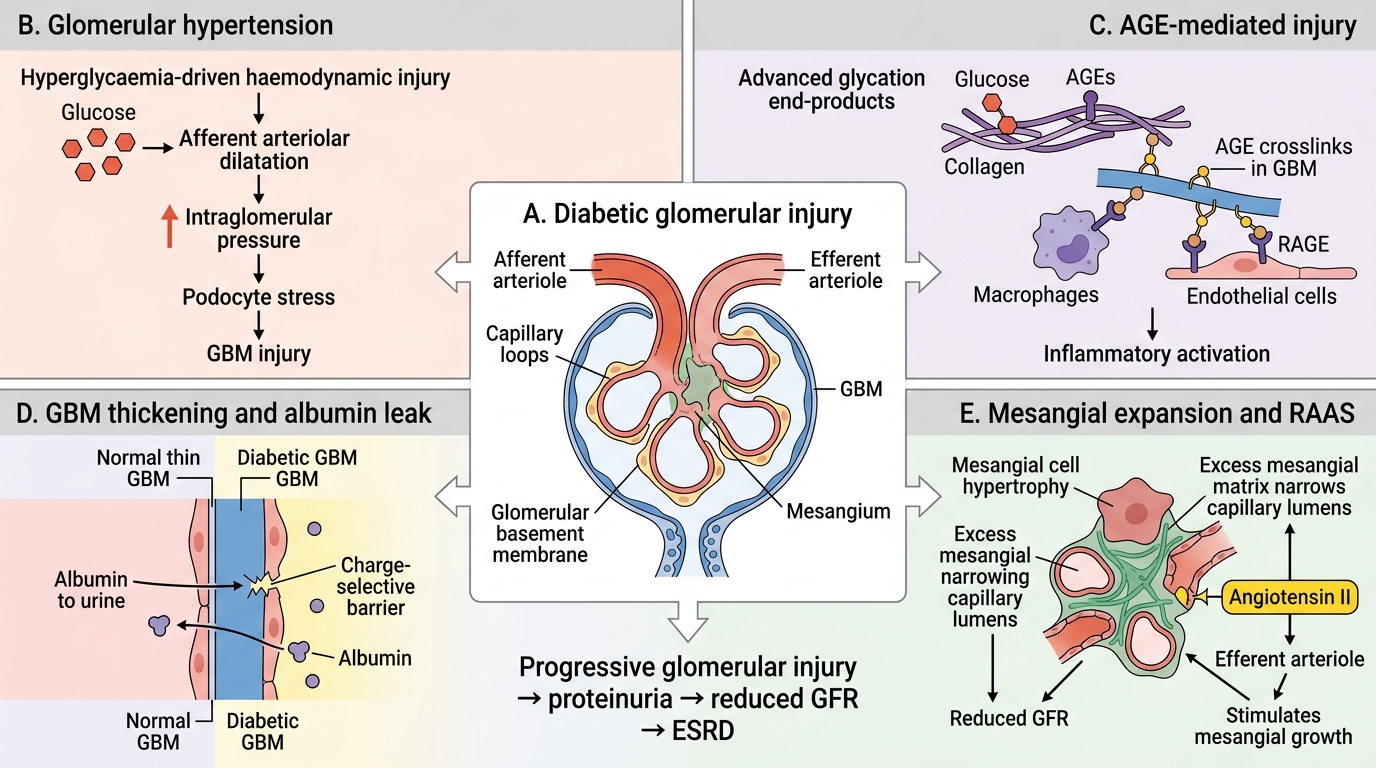
Pathogenesis of Diabetic Nephropathy
Diabetic nephropathy is the single most common cause of ESRD globally, complicating approximately 30–40% of patients with type 1 and type 2 DM over 20 years.
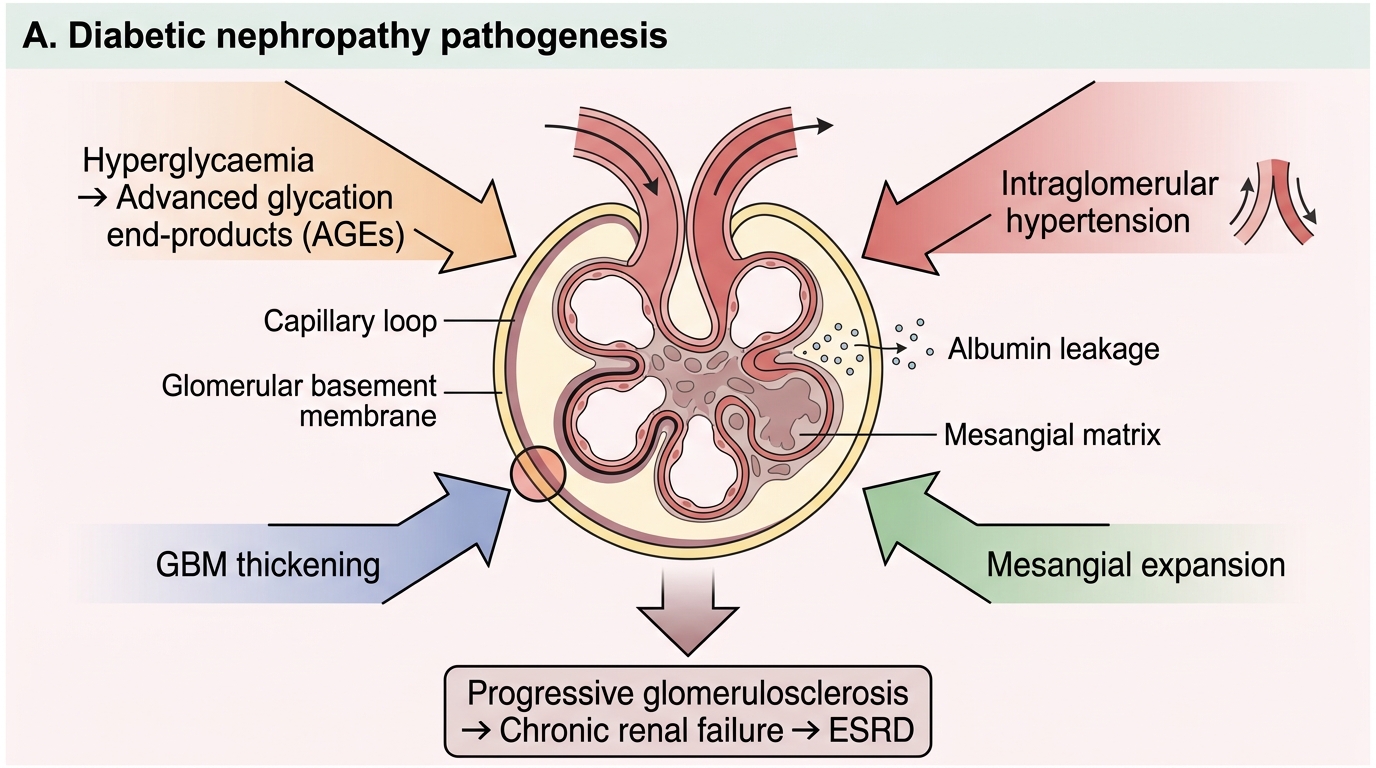
Mechanism of glomerular injury — four converging pathways:
- Hyperglycaemia-driven haemodynamic injury — sustained hyperglycaemia causes afferent arteriolar dilatation (via nitric oxide and prostaglandins), raising intraglomerular pressure (glomerular hypertension). This mechanical stress directly injures podocytes and the GBM.
- Advanced glycation end-products (AGEs) — glucose non-enzymatically glycates proteins. AGEs crosslink GBM collagen, increasing permeability, stimulate mesangial matrix production, and directly activate macrophages and endothelium via RAGE receptors.
- GBM thickening — the earliest ultrastructural change. Glycated collagen IV accumulates, the GBM widens, and its charge-selective barrier is disrupted → initially microalbuminuria.
- Mesangial expansion — AGE-driven mesangial cell hypertrophy and excess matrix deposition progressively narrows capillary lumens, reducing GFR.
Another amplifier: the activated renin-angiotensin system (angiotensin II is a direct mesangial mitogen and raises efferent arteriolar tone, worsening intraglomerular hypertension).
Pathogenesis of Diabetic Nephropathy
Diabetic Nephropathy — Morphology and Clinical Stages
Diabetic Nephropathy: Morphology and Clinical Stages
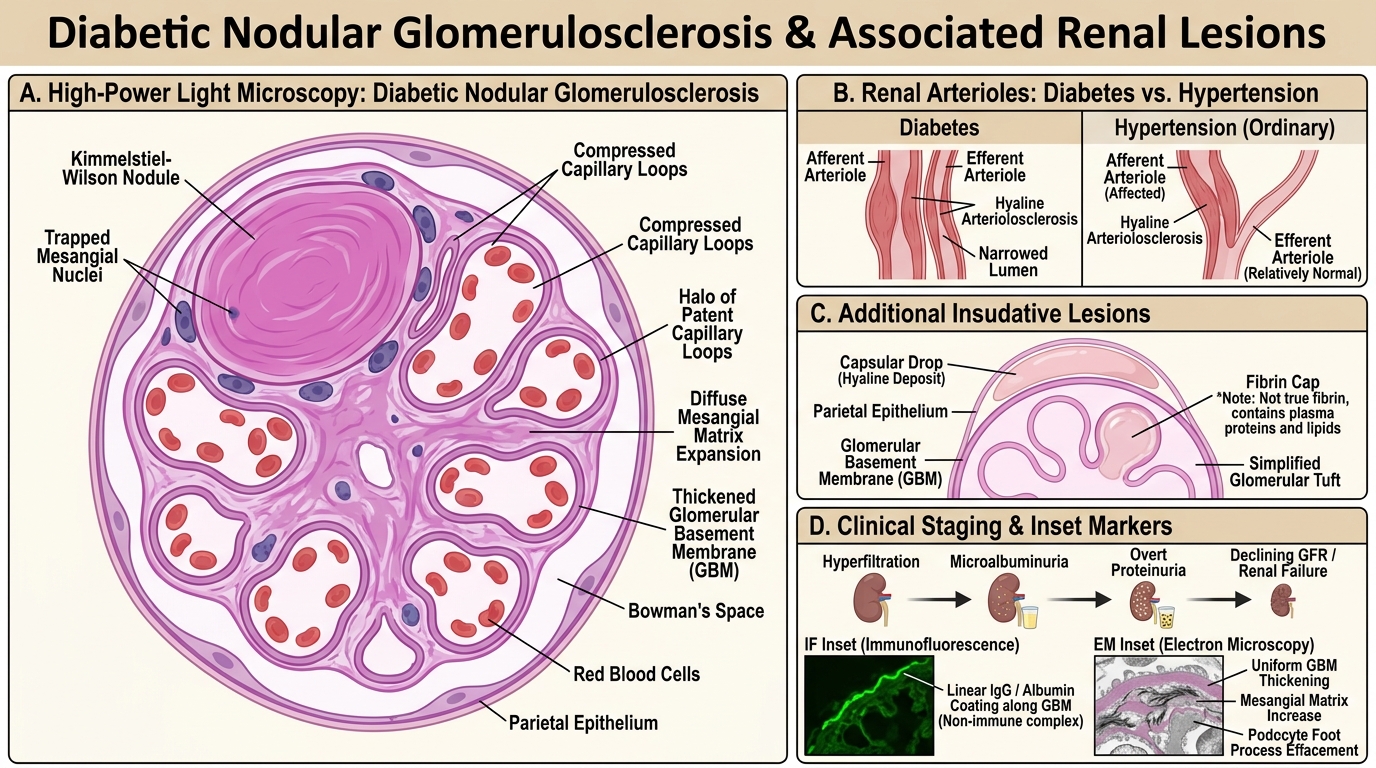
Light microscopy findings — two morphological patterns:
- Diffuse glomerulosclerosis — uniform increase in mesangial matrix and GBM thickening affecting ALL glomeruli. The more common pattern; alone it is not diagnostic of diabetes.
- Nodular glomerulosclerosis (Kimmelstiel-Wilson lesion) — pathognomonic for diabetic nephropathy. Ovoid PAS-positive acellular hyaline nodules sit at the periphery of the mesangium, expanding and compressing adjacent capillary loops. The nodule is laminated, contains trapped mesangial nuclei at the periphery, and is surrounded by patent capillaries (the "halo" of open loops). This pattern is seen in ~10–15% of diabetic biopsies.
Additional findings:
• Hyaline arteriolosclerosis — involving BOTH afferent and efferent arterioles (efferent involvement is virtually unique to diabetes; in ordinary hypertension, only afferent is affected).
• "Capsular drop" — hyaline deposit between GBM and parietal epithelium of Bowman's capsule.
• "Fibrin cap" — hyaline material occluding a glomerular capillary loop (misnomer; not fibrin).
• Tubular atrophy and interstitial fibrosis in advanced stages.
IF: Non-specific IgG and albumin linear coating along GBM ("insudative" due to leaky vessels — NOT immune complex).
EM: Uniform GBM thickening, mesangial matrix increase, preserved podocyte foot processes early → effacement late.
Clinical staging (Mogensen stages — simplified):
| Stage | Feature | Duration |
|---|---|---|
| 1 | Hyperfiltration, GFR ↑ | Years 1–5 |
| 2 | Silent; microstructural changes | Years 5–10 |
| 3 | Microalbuminuria 30–300 mg/day | Years 10–15 |
| 4 | Overt proteinuria >300 mg/day, GFR declining | Years 15–20 |
| 5 | ESRD | >20 years |
The microalbuminuria→overt proteinuria→ESRD trajectory is the most clinically important progression sequence to memorise.
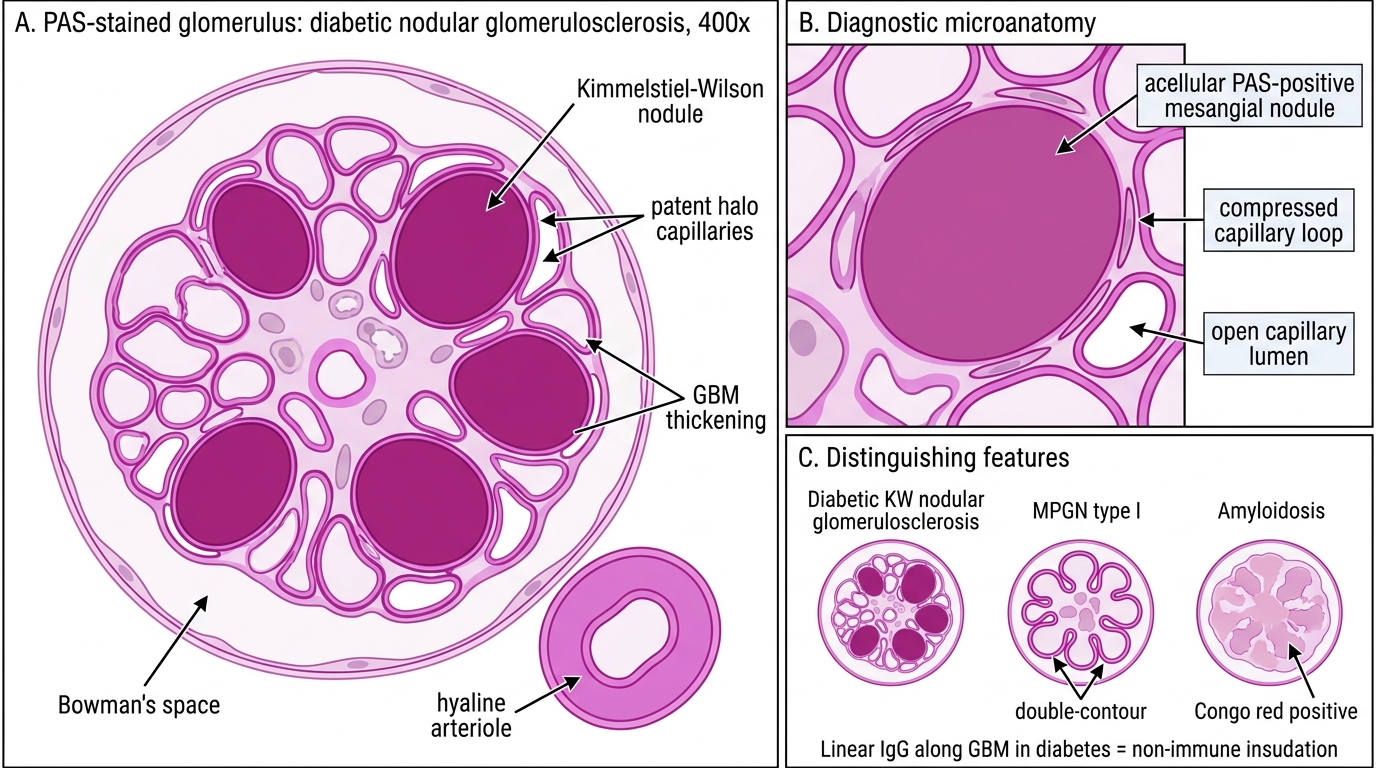
Kimmelstiel-Wilson Nodular Glomerulosclerosis
SELF-CHECK
A renal biopsy from a 58-year-old diabetic patient shows ovoid, PAS-positive acellular nodules at the periphery of the mesangium, surrounded by patent capillary loops. Immunofluorescence shows non-specific linear IgG along the GBM. What is the most specific diagnosis?
A. Membranoproliferative glomerulonephritis type I
B. Kimmelstiel-Wilson nodular glomerulosclerosis
C. AL amyloidosis with Congo-red positive deposits
D. Fibrillary glomerulonephritis
Reveal Answer
Answer: B. Kimmelstiel-Wilson nodular glomerulosclerosis
Kimmelstiel-Wilson (KW) nodular glomerulosclerosis is pathognomonic for diabetic nephropathy. The lesion is PAS-positive, acellular, located at mesangial periphery, and surrounded by a halo of patent capillary loops — distinguishing it from MPGN (lobular pattern, immune deposits, C3 positivity) and amyloid (Congo-red positive, amorphous, not PAS-bright in the same way). Linear IgG along GBM in diabetes is insudative (non-immune), not the granular pattern of immune complex diseases.
Lupus Nephritis — Immune-Complex Basis and ISN/RPS Classification
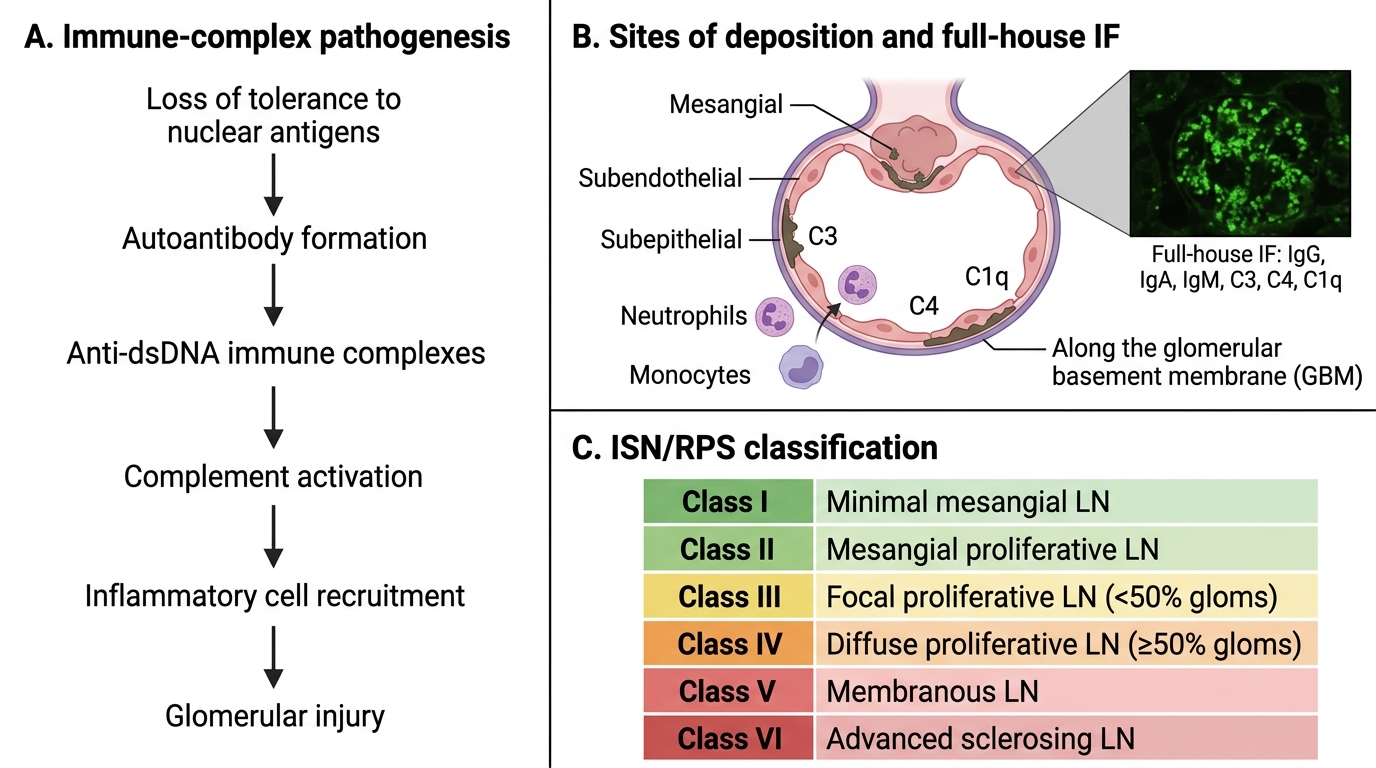
Lupus Nephritis: Immune-Complex Basis and ISN/RPS Classes
Lupus nephritis (LN) develops in 50–70% of patients with systemic lupus erythematosus (SLE). Renal involvement is the major determinant of morbidity and mortality in SLE.
Pathogenesis — Loss of tolerance to nuclear antigens (dsDNA, histones, Smith antigen) generates autoantibodies. Anti-dsDNA antibodies form immune complexes that deposit in glomeruli (mesangium, subendothelial space, subepithelial space, GBM), activating complement (C3, C4, C1q) and recruiting neutrophils and monocytes → glomerular inflammation.
IF hallmark: "full-house" immunofluorescence — simultaneous positivity for IgG, IgA, IgM, C3, C4, and C1q. This pattern is virtually diagnostic of lupus nephritis.
ISN/RPS classification (2003/2018) — six classes based on the site and extent of immune complex deposition and histological injury:
| Class | Name | Key Feature | Clinical |
|---|---|---|---|
| I | Minimal mesangial LN | Normal LM, mesangial deposits on IF/EM | Nil |
| II | Mesangial proliferative LN | Mesangial hypercellularity, mesangial deposits | Mild proteinuria/haematuria |
| III | Focal LN | <50% glomeruli involved, segmental/global endocapillary proliferation | Moderate nephritis |
| IV | Diffuse LN | ≥50% glomeruli, global or segmental lesions | Nephrotic+nephritic, severe |
| V | Membranous LN | Subepithelial deposits, GBM thickening | Heavy proteinuria, nephrotic |
| VI | Advanced sclerosing LN | ≥90% global glomerulosclerosis | CKD/ESRD |
Class IV (diffuse proliferative lupus nephritis) — the most severe class, and the most important to know:
• Endocapillary and mesangial proliferation in >50% of glomeruli.
• Wire-loop lesions — massive subendothelial immune complex deposits form a thickened, homogeneous, eosinophilic rim along capillary walls, resembling a wire loop on H&E and PAS.
• Hyaline thrombi (pseudothrombi) — large luminal immune complex masses.
• Crescents (fibrocellular), tubular atrophy in active severe disease.
• Full-house IF; "fingerprint" (tubuloreticular inclusion on EM, a type I interferon marker).
Class IV has the worst prognosis without treatment and the best response to aggressive immunosuppression (cyclophosphamide/mycophenolate + steroids).
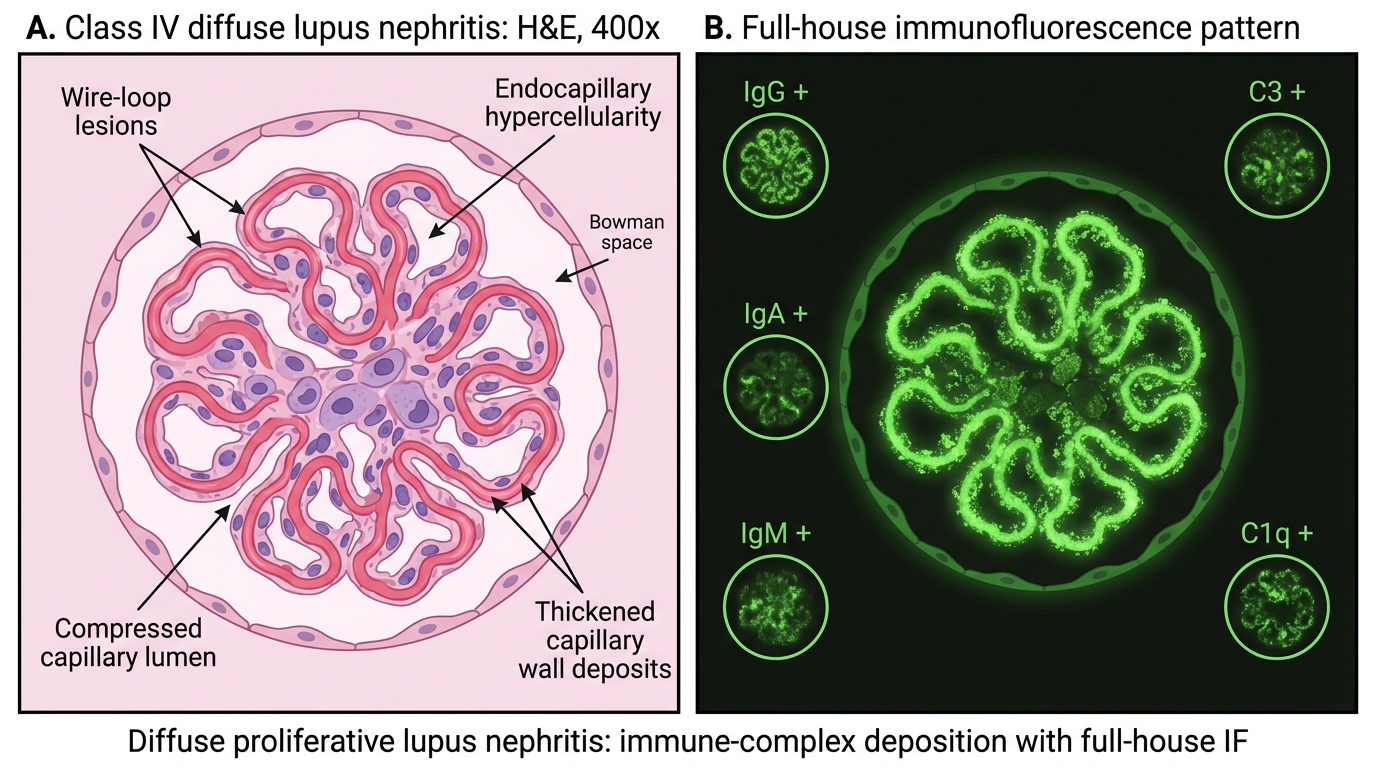
Class IV Lupus Nephritis: Wire-Loop Lesions and Full-House IF
CLINICAL PEARL
Class IV lupus nephritis and post-infectious GN can both produce endocapillary proliferation with "full-house" IF creating diagnostic confusion. The discriminators: (1) full-house IF (IgG+IgA+IgM+C3+C4+C1q together) strongly favours lupus — PIGN typically shows isolated IgG+C3 with predominant hump-shaped subepithelial deposits (EM). (2) Clinical context: ANA, anti-dsDNA, complement levels (low C3+C4 in lupus, often low C3 alone in PIGN). (3) Tubuloreticular inclusions on EM — type-I-interferon marker, essentially diagnostic for lupus. Pathology and serology must be read together.
SELF-CHECK
Renal biopsy immunofluorescence shows simultaneous positivity for IgG, IgA, IgM, C3, C4, and C1q in a 24-year-old woman with facial rash, arthralgia, and heavy proteinuria. What ISN/RPS class is MOST likely if more than 50% of glomeruli show global endocapillary proliferation on light microscopy?
A. Class II — mesangial proliferative lupus nephritis
B. Class III — focal lupus nephritis
C. Class IV — diffuse lupus nephritis
D. Class V — membranous lupus nephritis
Reveal Answer
Answer: C. Class IV — diffuse lupus nephritis
Class IV (diffuse lupus nephritis) is defined by involvement of ≥50% of glomeruli with global or segmental endocapillary proliferation, combined with the full-house IF pattern (IgG+IgA+IgM+C3+C4+C1q). Class III affects <50% of glomeruli. Class V is dominated by subepithelial deposits and GBM thickening (membranous pattern). Class II shows only mesangial changes on LM. The clinical setting (SLE, heavy proteinuria, full-house IF) and ≥50% glomerular involvement unambiguously indicate class IV.