Page 9 of 27
PA27.12-13 | Cystic Kidney Disease, Renal Stones & Obstructive Uropathy — SDL Guide
Learning Objectives
- Classify cystic kidney diseases and distinguish ADPKD from ARPKD on the basis of genetics, morphology, and clinical course.
- Explain the pathogenesis and extrarenal manifestations of autosomal dominant polycystic kidney disease (ADPKD).
- Identify the four major types of renal stones, their composition, causative conditions, and radiological characteristics.
- Describe the mechanism and morphological stages of hydronephrosis and the complications of obstructive uropathy.
INSTRUCTIONS
Cystic and obstructive diseases together account for a significant proportion of chronic kidney disease and transplant referrals in India. Understanding the genetic basis of PKD, the metabolic drivers of nephrolithiasis, and the pressure-atrophy cascade of obstruction provides the clinico-pathological framework you will use every time you interpret a renal biopsy report or counsel a patient with flank pain. Work through each section systematically; the micro-quizzes track your self-assessment at key branch points.
References
- Robbins & Kumar: Basic Pathology, 11th ed., Chapter 14 — The Kidney (textbook)
- Harsh Mohan: Textbook of Pathology, 8th ed., Chapter 22 — Kidney and Lower Urinary Tract (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 38-year-old man presents to the nephrology OPD with poorly controlled hypertension, bilateral flank fullness, and a family history of 'kidney problems' — his father died on dialysis. Ultrasound shows both kidneys massively enlarged and studded with cysts of varying sizes. Blood urea and creatinine are elevated. What single genetic mutation could have set this cascade in motion thirty years before his kidneys began to fail?
WHY THIS MATTERS
Cystic kidney disease encompasses a spectrum from the devastating ADPKD to benign simple cysts found incidentally on imaging. Distinguishing them determines whether you order genetic testing, screen family members, initiate antihypertensives early, or simply reassure. Renal stones affect roughly 12% of the Indian population during their lifetime, and obstructive uropathy is the third commonest cause of renal failure in children. The pathological mechanisms you learn here directly inform surgical urgency and long-term renal preservation strategies.
RECALL
Before reading on, spend two minutes answering these silently:
- What is the difference between autosomal dominant and autosomal recessive inheritance in terms of generations affected and carrier probability?
- What are the normal functions of the renal tubular epithelium in concentrating urine?
- Name two common causes of urinary tract obstruction you have encountered in surgery or medicine postings.
These anchor your Year-1 knowledge to the pathological detail ahead.
Classification of Cystic Kidney Disease
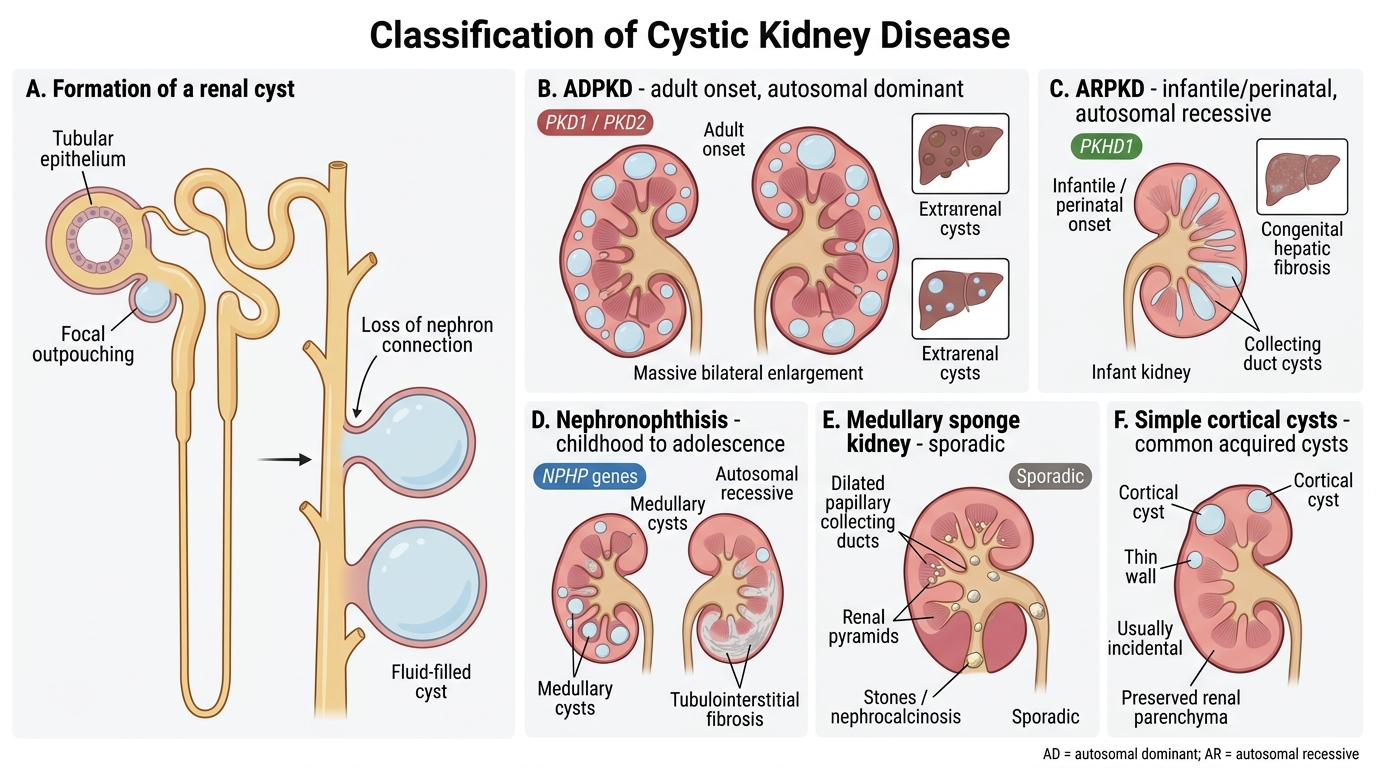
Classification of Cystic Kidney Disease
Renal cysts arise from focal outpouchings of tubular or collecting duct epithelium that lose connection with the parent nephron. A practical classification groups them by genetic basis, age of onset, and renal consequence:
| Disease | Inheritance | Onset | Key Feature |
|---|---|---|---|
| ADPKD | AD (PKD1/PKD2) | Adult | Bilateral massive enlargement, extrarenal cysts |
| ARPKD | AR (PKHD1) | Infantile/perinatal | Congenital hepatic fibrosis |
| Nephronophthisis | AR (NPHP genes) | Childhood–adolescence | Medullary cysts, tubulo-interstitial fibrosis |
| Medullary sponge kidney | Sporadic | Any | Dilated papillary collecting ducts, stones |
| Simple cortical cysts | Sporadic (somatic) | Middle age+ | Single/few, benign, very common |
| Acquired cystic disease | Acquired | End-stage / dialysis | Multiple bilateral cysts, ↑ RCC risk |
| Multicystic dysplastic kidney | Developmental | Neonatal | Non-functional, unilateral, mass |
Genetic (hereditary) cysts carry systemic implications; sporadic and acquired cysts are usually incidental findings.
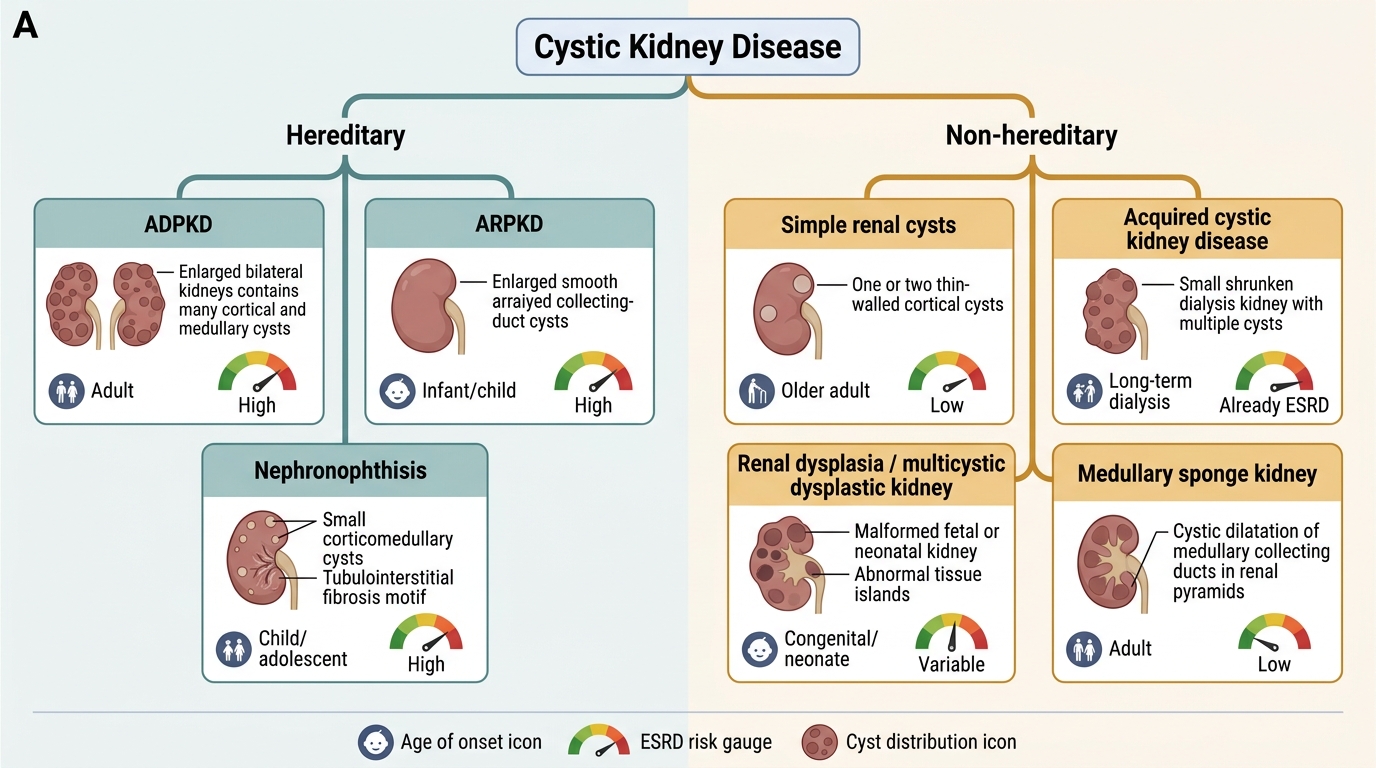
Classification of Cystic Kidney Disease
ADPKD — Genetics and Molecular Pathogenesis
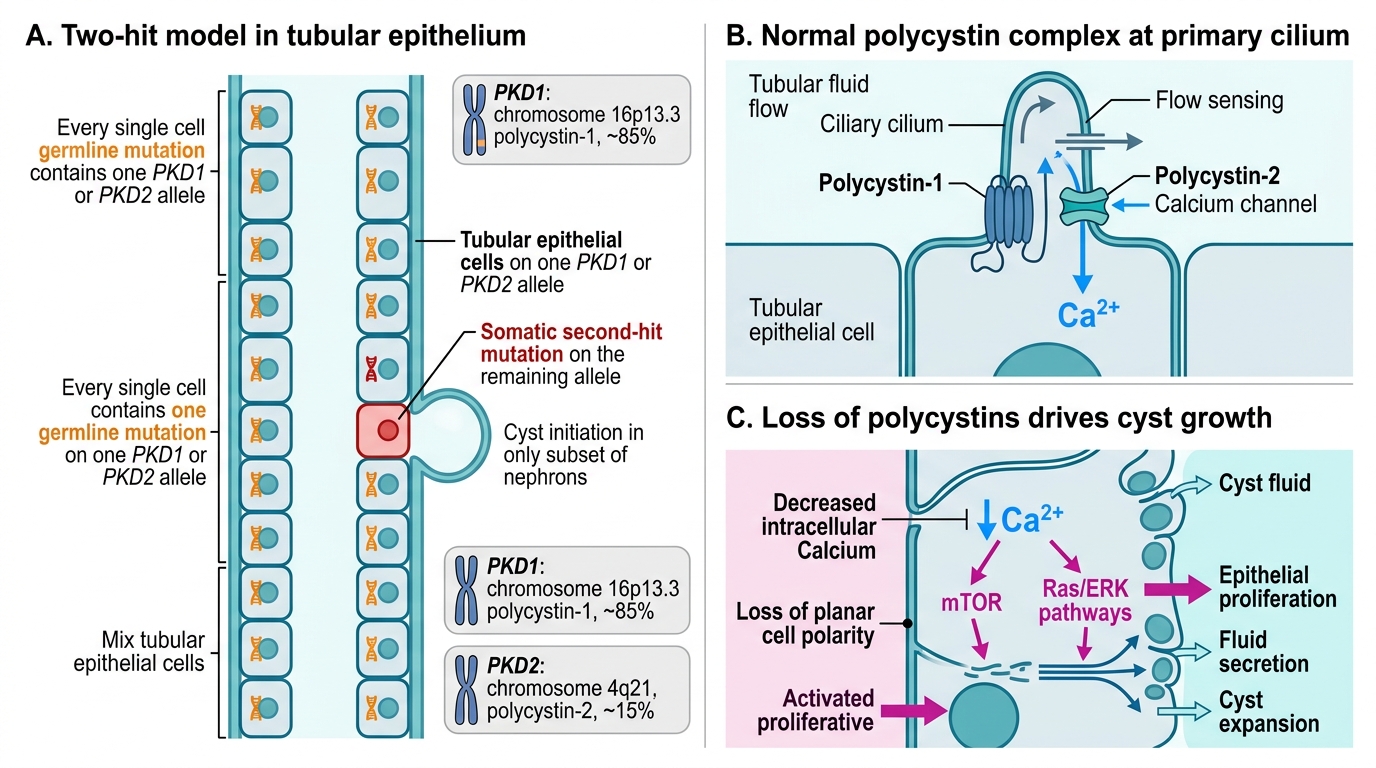
ADPKD: Two-Hit Molecular Pathogenesis
Autosomal dominant polycystic kidney disease (ADPKD) is the commonest inherited kidney disease, affecting approximately 1 in 1,000 individuals worldwide.
Genetic basis — two genes:
- PKD1 (chromosome 16p13.3) — encodes polycystin-1, a large transmembrane receptor. Mutated in ~85% of cases. Associated with earlier, more severe disease (ESRD by ~54 years).
- PKD2 (chromosome 4q21) — encodes polycystin-2, a calcium-channel protein. Mutated in ~15% of cases. Milder course (ESRD by ~74 years).
Polycystin-1 and polycystin-2 form a complex localised to primary cilia of tubular epithelial cells. Their function is to transduce mechanical flow-sensing signals into intracellular calcium flux. Loss of this complex activates proliferative pathways (mTOR, Ras/ERK) and impairs cell polarity, driving cyst initiation.
Two-hit model: although ADPKD is dominant (one germline mutation is sufficient to cause disease), somatic mutation of the second allele in individual tubular cells triggers cyst formation in those cells specifically — explaining why only a subset of nephrons cyst-form despite every cell carrying the germline mutation.
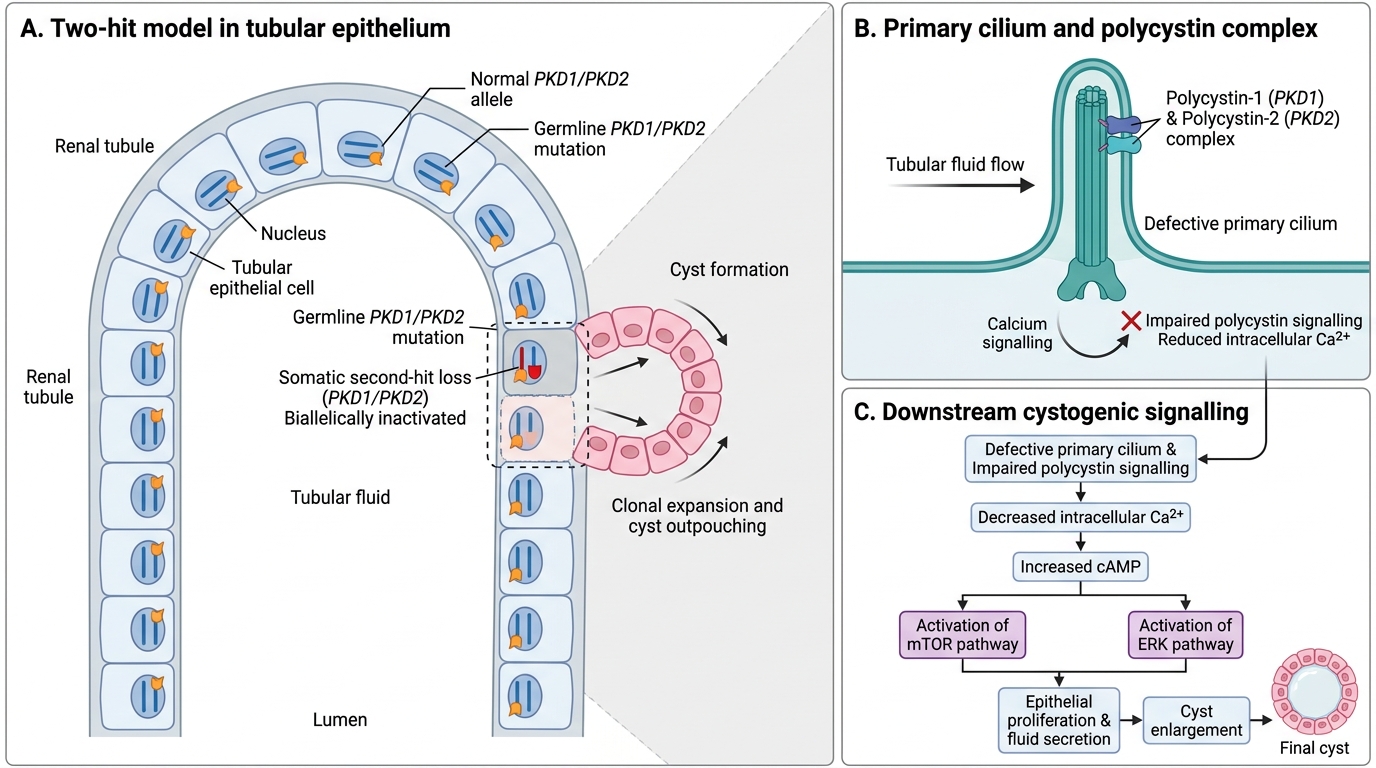
Two-Hit Model of ADPKD Cyst Formation
ADPKD — Morphology and Extrarenal Manifestations
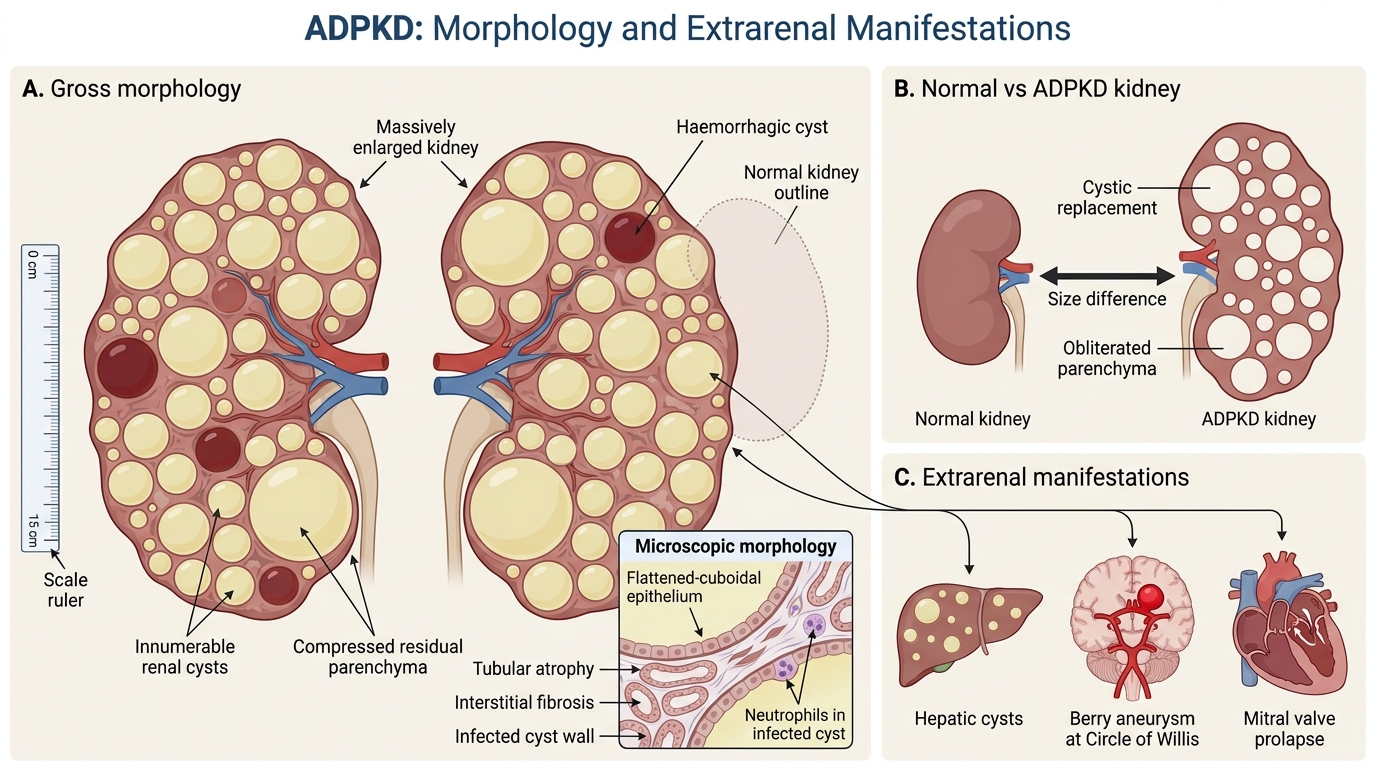
ADPKD: Morphology and Extrarenal Manifestations
Gross morphology: Both kidneys are massively enlarged — combined weight may exceed 4 kg (normal combined ~300 g). The external surface is replaced by hundreds of cysts ranging from a few millimetres to several centimetres, filled with clear, serous, or haemorrhagic fluid. Normal parenchyma is progressively obliterated.
Microscopic morphology: Cyst walls are lined by a flattened to cuboidal epithelium. The residual nephrons show tubular atrophy and interstitial fibrosis as compression progresses. Secondary infection may cause neutrophilic infiltration (infected cysts).
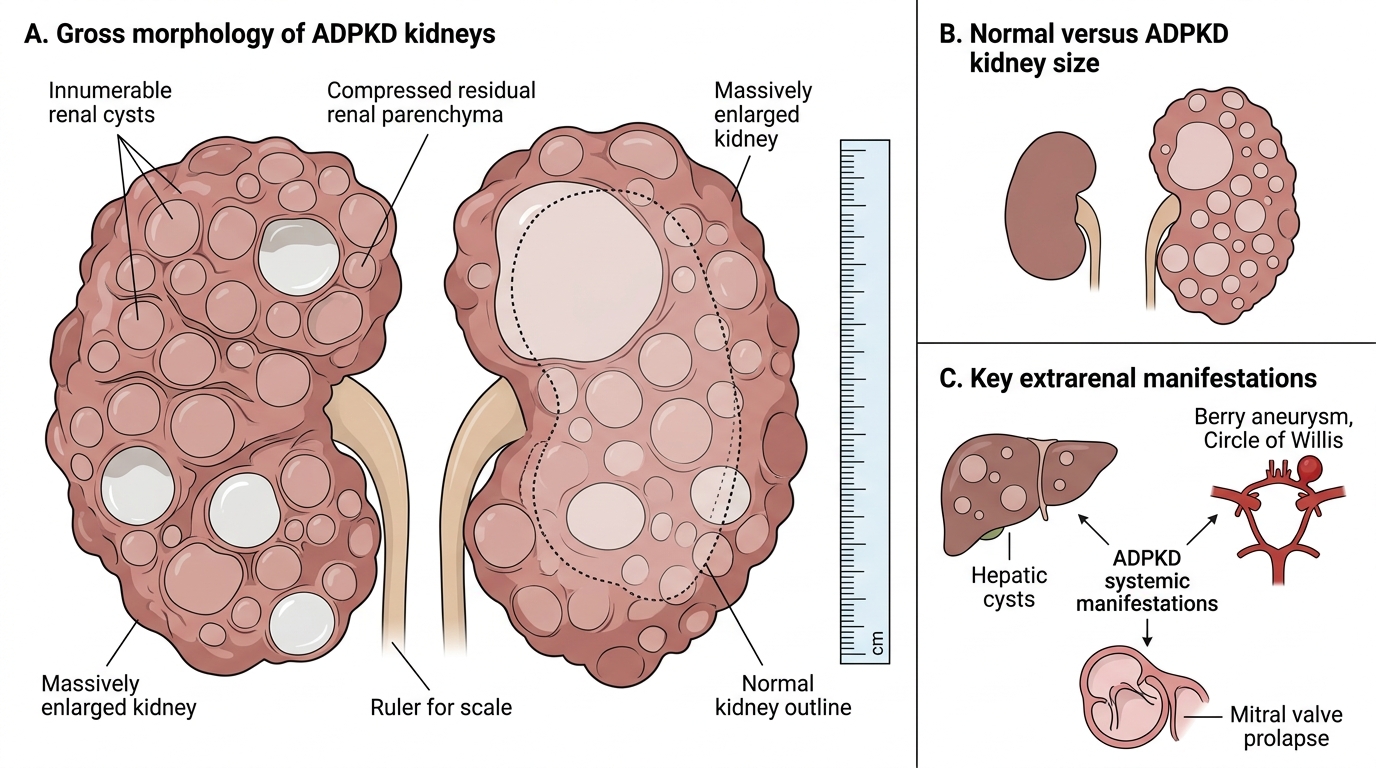
Gross Morphology and Systemic Manifestations of ADPKD
Extrarenal (systemic) manifestations — critical for exams and clinical practice:
- Hepatic cysts — in ~75% of patients; usually non-functional; the most common extrarenal manifestation.
- Berry (saccular) aneurysms — in ~10–15%; arise at branch points of the Circle of Willis; rupture causes subarachnoid haemorrhage (SAH) — the most lethal extrarenal complication.
- Mitral valve prolapse (MVP) — in ~25%; generally haemodynamically insignificant.
- Intracranial cysts, pancreatic cysts, seminal vesicle cysts (less common).
Clinical course: Patients are typically asymptomatic until the 4th–5th decade. Presenting features include hypertension (renin–angiotensin activation as cysts compress intrarenal vessels), gross haematuria (cyst rupture), flank pain, and palpable kidneys. Proteinuria is mild. Progression to end-stage renal disease (ESRD) occurs in ~50% by age 60.
SELF-CHECK
A 42-year-old man with ADPKD presents with the sudden onset of the worst headache of his life. CT shows blood in the subarachnoid space. The responsible extrarenal lesion is:
A. Hepatic cyst rupture
B. Berry aneurysm rupture at the Circle of Willis
C. Mitral valve vegetations embolising to cerebral vessels
D. Renal vein thrombosis with paradoxical embolism
Reveal Answer
Answer: B. Berry aneurysm rupture at the Circle of Willis
Berry (saccular) aneurysms at Circle of Willis branch-points are the most lethal extrarenal manifestation of ADPKD. Their rupture produces classic thunderclap headache and subarachnoid haemorrhage. Hepatic cysts are common but do not cause intracranial bleeding. MVP in ADPKD is haemodynamically benign. Renal vein thrombosis does not directly cause SAH.