Page 10 of 27
PA27.12-13 | Cystic Kidney Disease, Renal Stones & Obstructive Uropathy — SDL Guide (Part 2)
ARPKD, Nephronophthisis, and Other Cystic Diseases
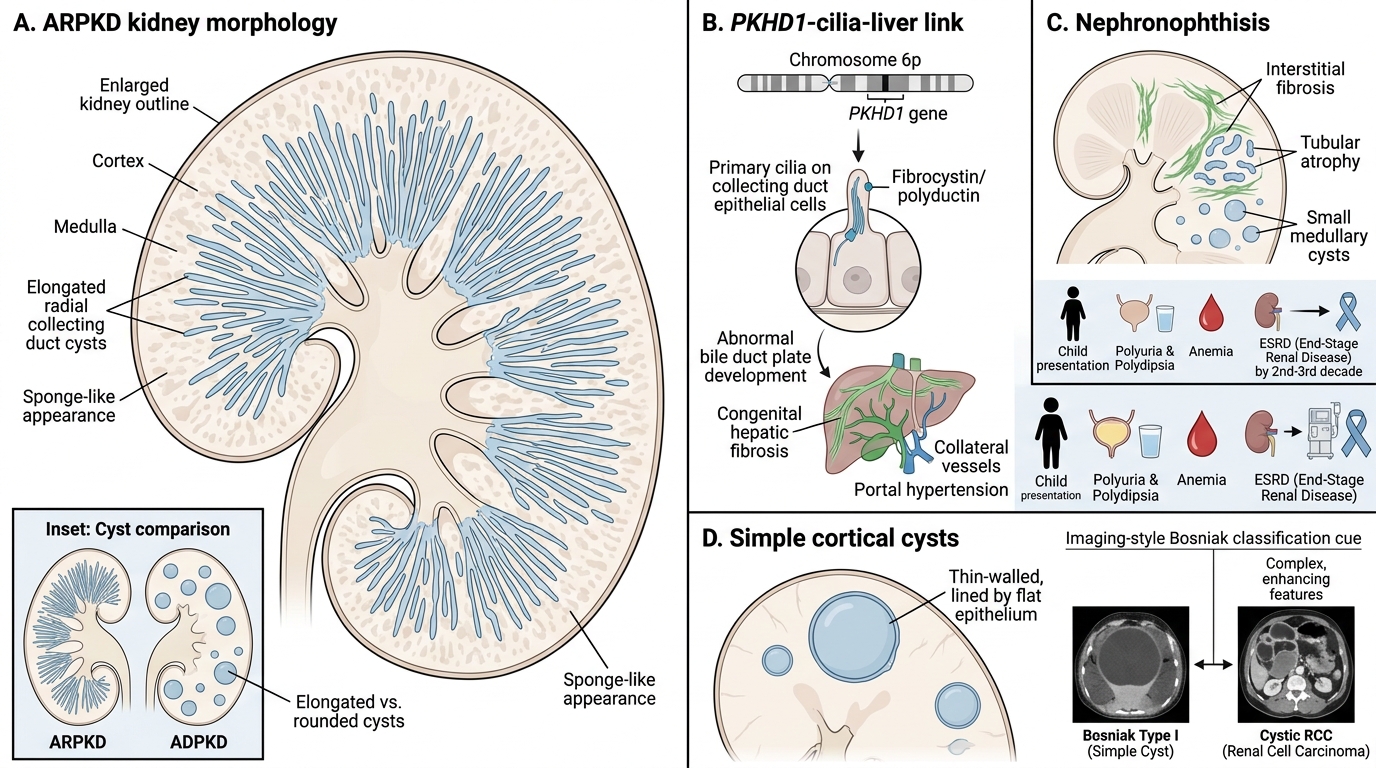
Cystic Diseases of the Kidney: ARPKD, Nephronophthisis, and Simple Cortical Cysts
Autosomal recessive PKD (ARPKD): Caused by mutations in PKHD1 (chromosome 6p) encoding fibrocystin/polyductin, a protein also localised to primary cilia. Disease manifests perinatally or in infancy. Kidneys are symmetrically enlarged with innumerable tiny elongated cysts arranged radially in the cortex and medulla — produced by dilation of collecting ducts. Unlike ADPKD cysts (which are rounded), ARPKD cysts are sponge-like on cut section. A hallmark association is congenital hepatic fibrosis (bile duct plate malformation), which leads to portal hypertension. Severe perinatal disease causes oligohydramnios and Potter sequence (pulmonary hypoplasia, facial deformities, limb deformities).
Nephronophthisis (NPH) / medullary cystic disease: A family of autosomal recessive tubulointerstitial nephropathies caused by mutations in NPHP genes (nephrocystins). Histology shows tubular atrophy, interstitial fibrosis, and small medullary cysts. Clinically presents in childhood with polyuria and polydipsia (tubular concentrating defect), anaemia, and progression to ESRD by the 2nd–3rd decade. NPH is the commonest genetic cause of ESRD in children.
Simple cortical cysts: The commonest renal cystic lesion (found in >50% of adults >50 years on imaging). Arise from somatic tubular dilation. Single or multiple, unilateral or bilateral; lined by flat epithelium. No genetic or malignant potential. Distinguished from cystic RCC by Bosniak imaging criteria.
Acquired cystic disease: Develops in patients on long-term dialysis (~90% after 10 years). Multiple bilateral cortical/medullary cysts. Clinically relevant because of a 50-fold increased risk of renal cell carcinoma arising within these cysts.
CLINICAL PEARL
Remember: 'It runs in the family but skips a generation' does NOT fit ADPKD. Every child of an affected parent has a 50% chance of inheriting the mutant allele. Apparent skipping is almost always late/undiagnosed disease or death from another cause before ESRD. Genetic counselling should be offered to all first-degree relatives of ADPKD patients.
SELF-CHECK
A neonate is found to have bilaterally enlarged kidneys with a sponge-like cut surface and features of Potter sequence. Which associated hepatic lesion is characteristically present?
A. Hepatocellular carcinoma
B. Hepatic adenoma
C. Congenital hepatic fibrosis
D. Polycystic liver disease identical to ADPKD
Reveal Answer
Answer: C. Congenital hepatic fibrosis
ARPKD (caused by PKHD1 mutations) is characteristically associated with congenital hepatic fibrosis — a bile duct plate malformation that leads to portal hypertension and hepatosplenomegaly. The polycystic liver disease seen in some ADPKD patients is a separate entity (hepatic cysts, not fibrosis) and does not cause portal hypertension. HCC and adenoma are not part of the ARPKD phenotype.
Renal Stones — Types, Pathogenesis, and Risk Factors
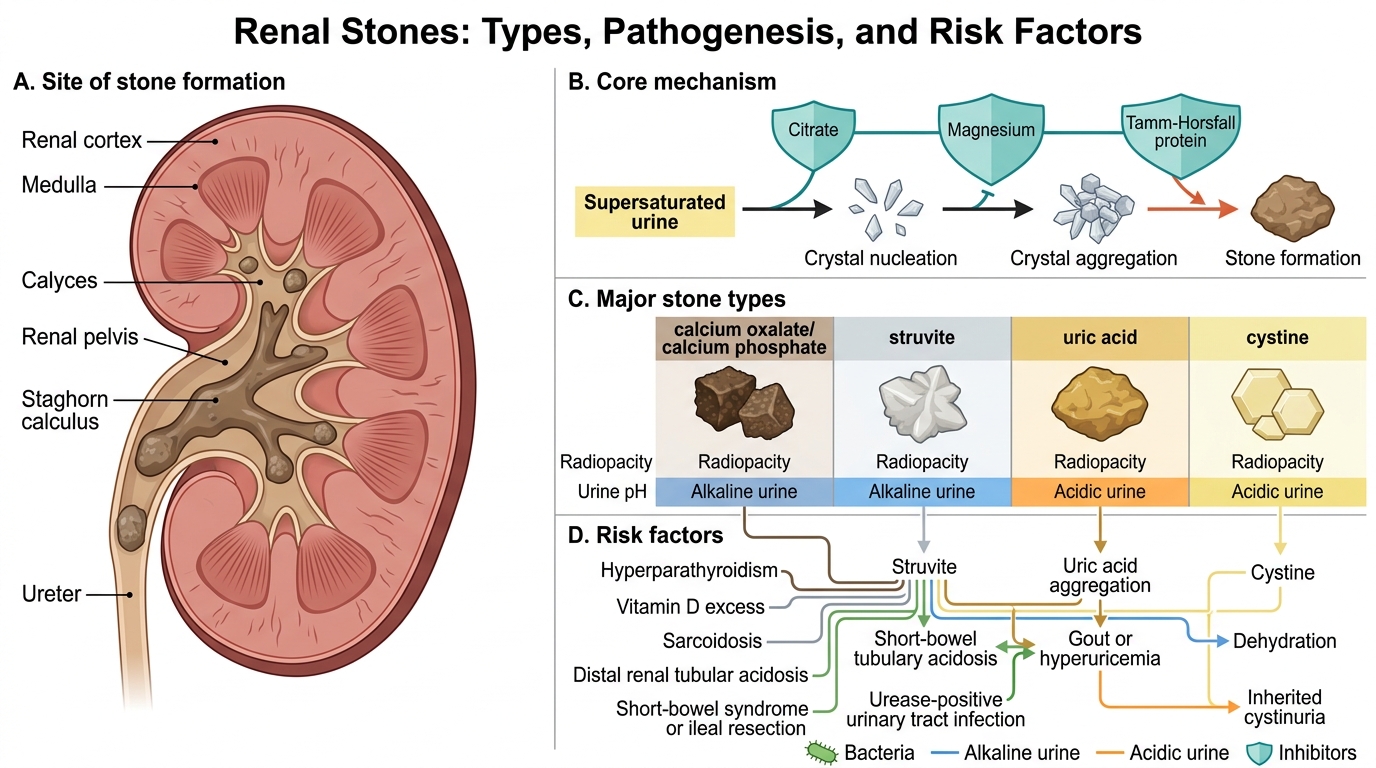
Renal Stones: Types, Pathogenesis, and Risk Factors
Nephrolithiasis (urolithiasis) refers to the formation of concretions (calculi) within the renal collecting system. Stones form when the urine is supersaturated with insoluble salts and inhibitory mechanisms (citrate, magnesium, Tamm–Horsfall protein) are overwhelmed.
Calcium oxalate / calcium phosphate stones
- Composition: 70–80% of all stones (commonest).
- Appearance: Hard, brown, rough; often form stag-horn configurations when large.
- Radio-opacity: Radiopaque (visible on plain X-ray).
- Pathogenesis: Requires hypercalciuria (absorptive, resorptive, or renal-leak type), hyperoxaluria (primary enzyme defect or absorptive — classically after ileal resection with increased colonic oxalate absorption), or hypocitraturia.
- Risk conditions: Hyperparathyroidism, vitamin D excess, sarcoidosis, distal RTA, short-bowel syndrome.
Struvite (triple-phosphate / magnesium ammonium phosphate) stones
- Composition: ~10–15% of stones.
- Appearance: Pale, soft, often grow to fill the renal pelvis → staghorn calculus.
- Radio-opacity: Radiopaque (mildly).
- Pathogenesis: Urease-producing bacteria (Proteus mirabilis, Klebsiella, Pseudomonas) split urea → ammonia → alkaline urine (pH >7.2) → struvite precipitation.
- Clinical significance: Indicate persistent UTI; difficult to eradicate without complete stone removal.
Uric acid stones
- Composition: ~5–10% of stones.
- Appearance: Yellow-brown, smooth.
- Radio-opacity: Radiolucent (not visible on plain X-ray; detected by ultrasound or CT).
- Pathogenesis: Hyperuricosuria (gout, high purine diet, myeloproliferative disease, tumour lysis) + persistently acidic urine (pH <5.5) favours uric acid precipitation (pKa 5.5).
- Treatment: Urinary alkalinisation with potassium citrate can dissolve existing stones.
Cystine stones
- Composition: ~1–2% of stones.
- Appearance: Yellow, waxy, hexagonal crystals on urine microscopy.
- Radio-opacity: Mildly radiopaque (cystine has some sulphur).
- Pathogenesis: Cystinuria — autosomal recessive defect in the SLC3A1/SLC7A9 dibasic amino acid transporter in the proximal tubule and jejunum, leading to excess cystine excretion. Cystine has very low solubility.
- Diagnostic clue: Hexagonal crystals on urine microscopy + family history of recurrent stones from childhood.
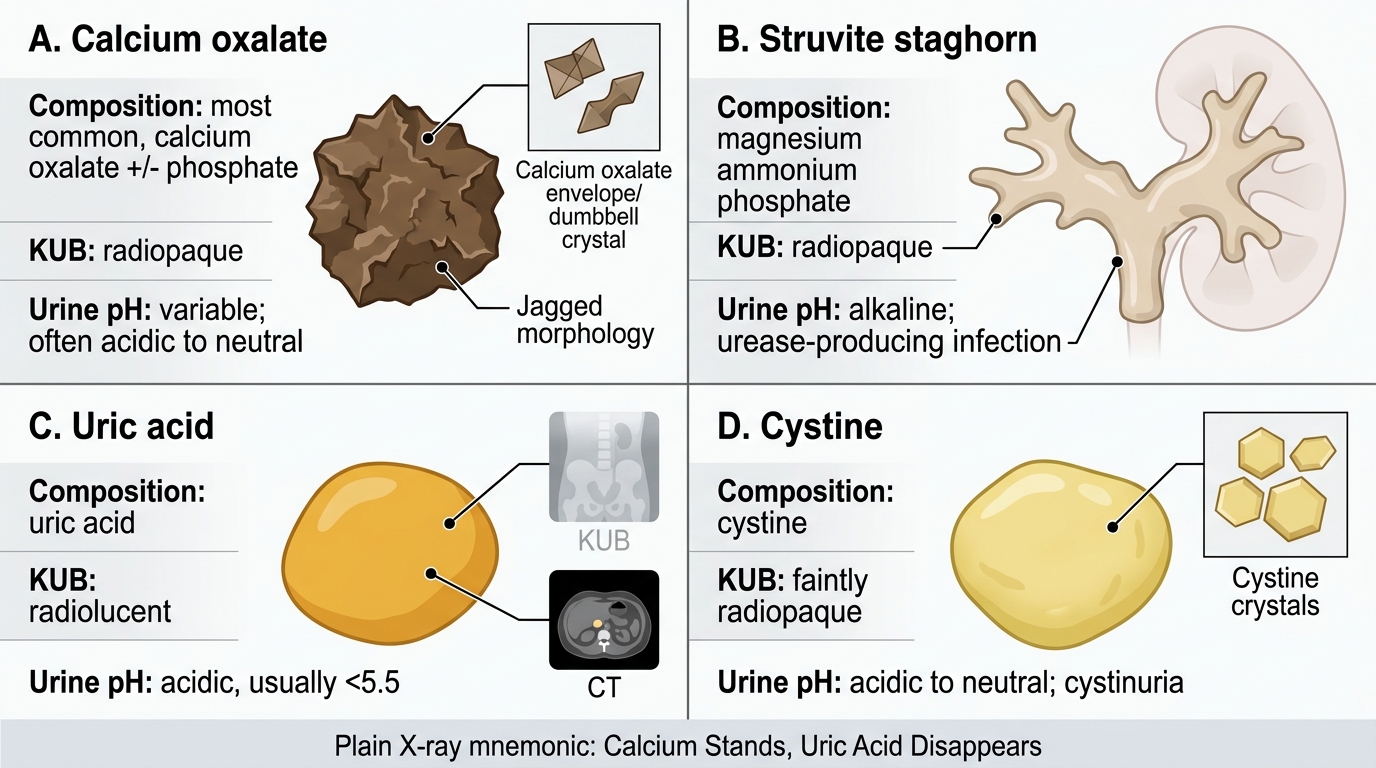
Major Renal Stone Types: Morphology, Opacity, and Urine pH
CLINICAL PEARL
Plain X-ray mnemonic for stone radio-opacity: 'Calcium Stands (radiopaque), Uric Acid Disappears (radiolucent)' — calcium oxalate, calcium phosphate, and struvite stones cast shadows on KUB; uric acid stones are invisible on plain film but evident on non-contrast CT. Cystine stones are faintly opaque. This distinction directs the initial imaging workup and monitors treatment response.
SELF-CHECK
A 28-year-old man with a history of gout presents with renal colic. Urine pH is 4.9. KUB X-ray shows NO stone. Which stone type is most likely, and why?
A. Calcium oxalate — radiopaque, would be visible
B. Struvite — alkaline urine favours formation
C. Uric acid — radiolucent, persistently acidic urine favours formation
D. Cystine — autosomal recessive, presents in childhood
Reveal Answer
Answer: C. Uric acid — radiolucent, persistently acidic urine favours formation
Uric acid stones are radiolucent and therefore invisible on plain KUB X-ray. Gout (hyperuricosuria) combined with persistently acidic urine (pH <5.5) is the classic setting. Struvite stones require alkaline urine from urease-producing bacteria — the opposite of this patient's pH. Calcium oxalate is radiopaque and would be visible. Cystine stones present from childhood with a family history, not typically in adult gout.
Obstructive Uropathy and Hydronephrosis — Pathogenesis
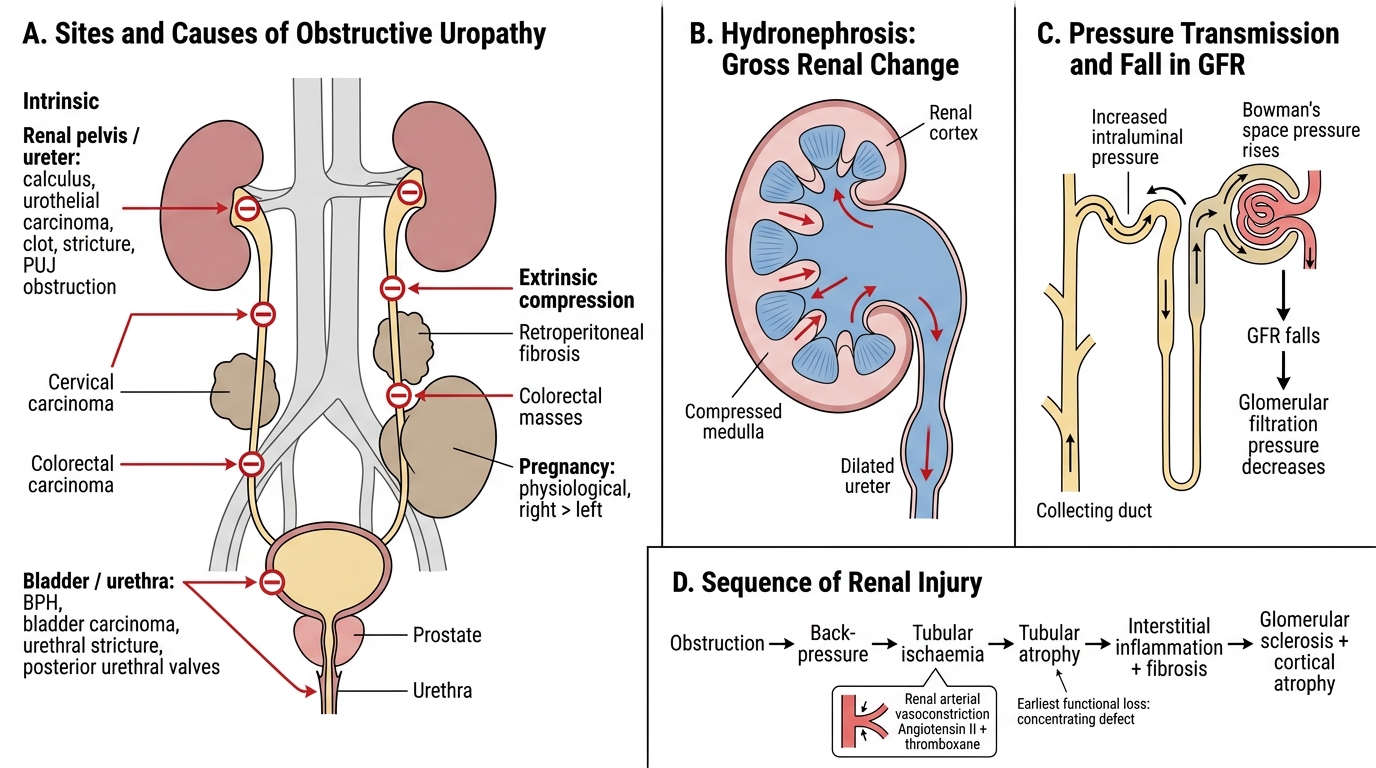
Obstructive Uropathy and Hydronephrosis: Pathogenesis
Obstructive uropathy refers to structural or functional obstruction to urine flow at any level — from the collecting tubule to the urethra. Hydronephrosis is the resulting dilation of the renal pelvis and calyces secondary to back-pressure.
Major causes by site:
Intrinsic (within the urinary tract):
- Renal pelvis/ureter: Calculi (commonest in adults), urothelial carcinoma, blood clots, strictures, pelvi-ureteric junction (PUJ) obstruction (commonest cause of hydronephrosis in children — often congenital aperistaltic segment).
- Bladder/urethra: Benign prostatic hyperplasia (BPH — commonest cause of bilateral obstruction in older males), bladder carcinoma, urethral stricture, posterior urethral valves (boys).
Extrinsic compression:
- Cervical carcinoma, colorectal carcinoma, retroperitoneal fibrosis, pregnancy (physiological, right > left).
Pathogenesis of renal injury:
- Back-pressure → increased intraluminal pressure in the collecting system.
- Pressure transmitted retrograde → proximal tubules → Bowman's space → glomerular filtration pressure decreases → GFR falls.
- Tubular ischaemia → tubular atrophy (concentrating defect is the earliest functional loss).
- Renal arterial vasoconstriction (angiotensin II, thromboxane) worsens ischaemia.
- Interstitial inflammation and progressive fibrosis → irreversible parenchymal atrophy.
The sequence: obstruction → back-pressure → ischaemia → tubular atrophy → glomerular sclerosis → cortical thinning → shrunken, scarred kidney.
Reversibility: Early obstruction (days–weeks) is reversible with relief. Prolonged obstruction (months) causes irreversible damage proportional to degree and duration.