Page 7 of 27
PA27.{11,15} | Vascular Diseases & Thrombotic Microangiopathies — SDL Guide (Part 3)
Thrombotic Thrombocytopenic Purpura (TTP)
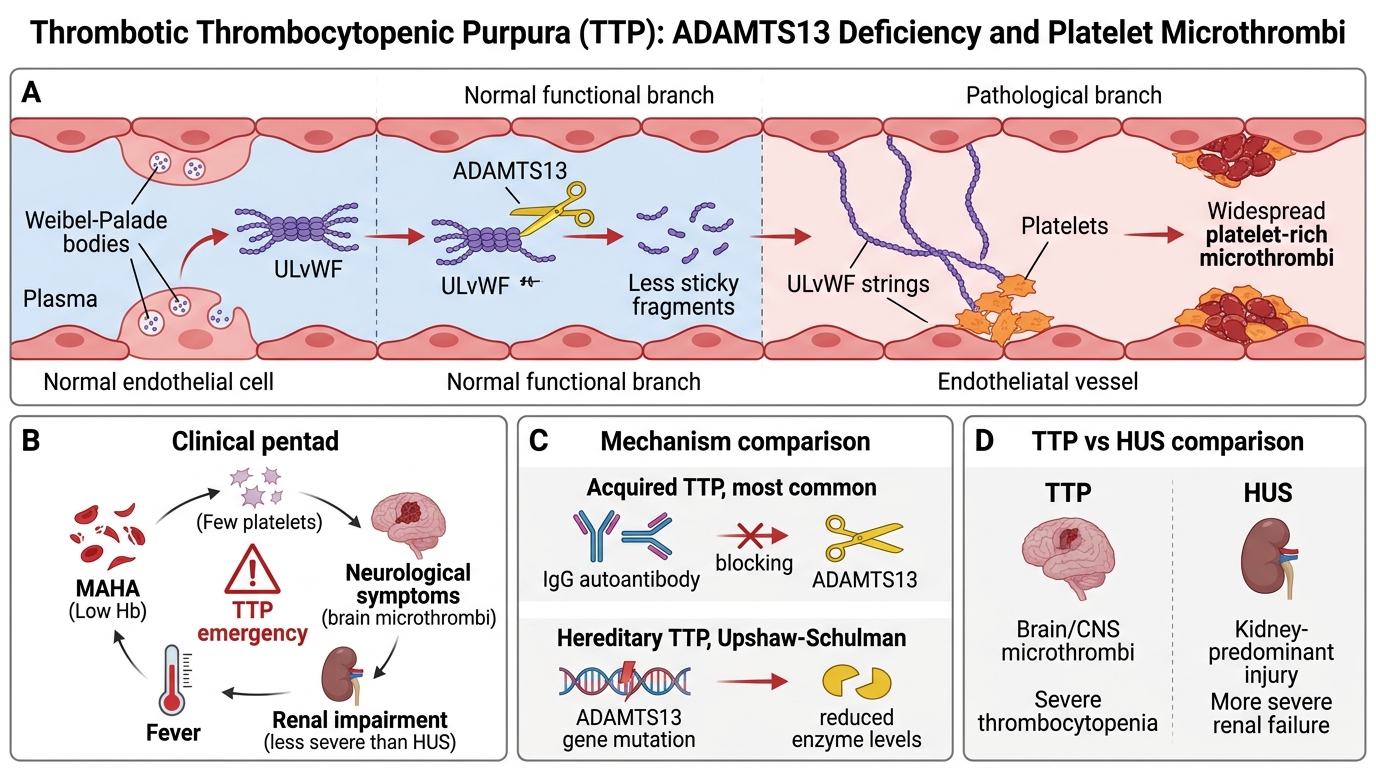
Thrombotic Thrombocytopenic Purpura: ADAMTS13 Deficiency
TTP is the most dangerous of the thrombotic microangiopathies, with historically ~90% mortality if untreated.
Pathogenesis — the ADAMTS13 story:
• Von Willebrand factor (vWF) is released as ultra-large multimers (ULvWF) from endothelial Weibel-Palade bodies; these are pro-thrombotic
• ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif, member 13) — a plasma metalloprotease — normally cleaves ULvWF into smaller, less sticky monomers
• ADAMTS13 deficiency → ULvWF accumulates → platelet aggregation on endothelial surfaces → widespread platelet-rich microthrombi
| Type | Mechanism |
|---|---|
| Acquired TTP (most common) | Autoantibody (IgG) against ADAMTS13 — inhibits cleavage activity |
| Hereditary TTP (Upshaw-Schulman) | ADAMTS13 gene mutation — rare, recurrent |
Clinical pentad of TTP (memorise):
1. MAHA (schistocytes, low Hb)
2. Thrombocytopenia (<30,000/μL — typically severe)
3. Neurological symptoms (fluctuating — confusion, seizures, focal deficits; from cerebral microthrombi)
4. Fever
5. Renal impairment (less severe than HUS — kidneys are not the primary target)
TTP vs HUS — key distinguishing features:
| Feature | TTP | HUS (typical) |
|---|---|---|
| Age | Adults (any age) | Children <5 |
| Prodrome | None (acquired) | Bloody diarrhoea |
| Neurological features | Prominent | Absent/mild |
| Renal failure | Mild-moderate | Severe (main feature) |
| ADAMTS13 | Severely low (<10%) | Normal |
| Platelet count | Very low (<30K) | Low (<80K) |
| Treatment | Plasma exchange (PLEX) | Supportive; eculizumab for aHUS |
Treatment of acquired TTP:
• Plasma exchange (PLEX/plasmapheresis) — removes anti-ADAMTS13 antibodies AND replaces ADAMTS13 enzyme. Dramatically reduces mortality from ~90% to <20%.
• Caplacizumab (anti-vWF nanobody) — newer adjunct
• Rituximab for refractory/relapsing cases
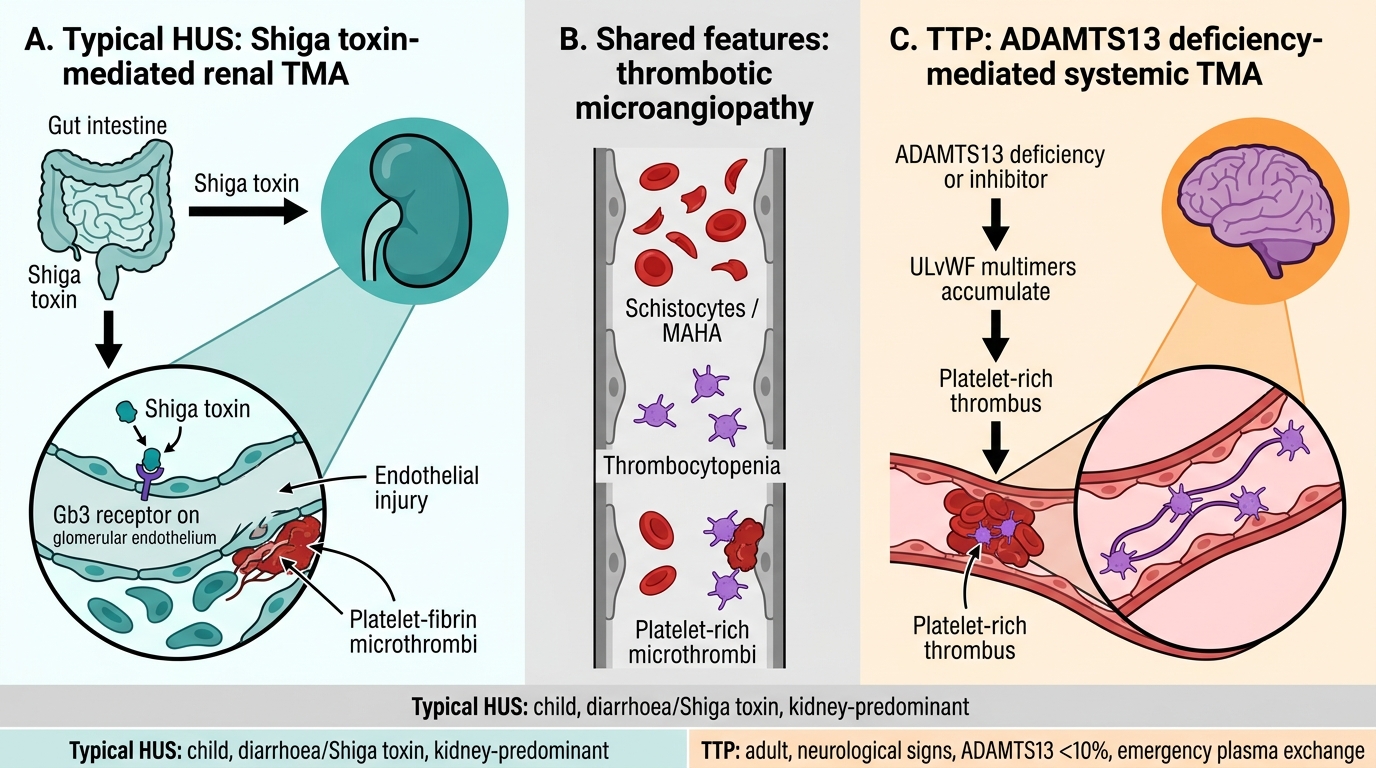
Typical HUS vs TTP: Mechanisms and Dominant Organ Targets
SELF-CHECK
A 34-year-old woman presents with confusion, fever, petechiae, and MAHA. ADAMTS13 activity is 4% (reference >67%). Shiga-toxin stool PCR is negative. Which treatment is the MOST critical immediate intervention?
A. Plasma exchange (plasmapheresis) to replace ADAMTS13 and remove inhibitor antibodies
B. Platelet transfusion to raise count above 50,000/μL
C. Eculizumab infusion to block terminal complement
D. Haemodialysis for acute kidney injury
Reveal Answer
Answer: A. Plasma exchange (plasmapheresis) to replace ADAMTS13 and remove inhibitor antibodies
This presentation — adult, no diarrhoea, prominent neurological features, very low ADAMTS13 (<10%) — is acquired TTP. Plasma exchange (PLEX) is the cornerstone emergency treatment: it simultaneously removes the autoantibody inhibiting ADAMTS13 and replenishes functional ADAMTS13 enzyme, reducing TTP mortality from ~90% to <20%. Platelet transfusion is CONTRAINDICATED in TTP — it adds fuel to microthrombus formation and has caused deaths. Eculizumab is for complement-mediated aHUS, not TTP. Haemodialysis may be needed but is not the primary TTP intervention.
DIC, Scleroderma Renal Crisis, Cortical Necrosis, and Renal Infarction
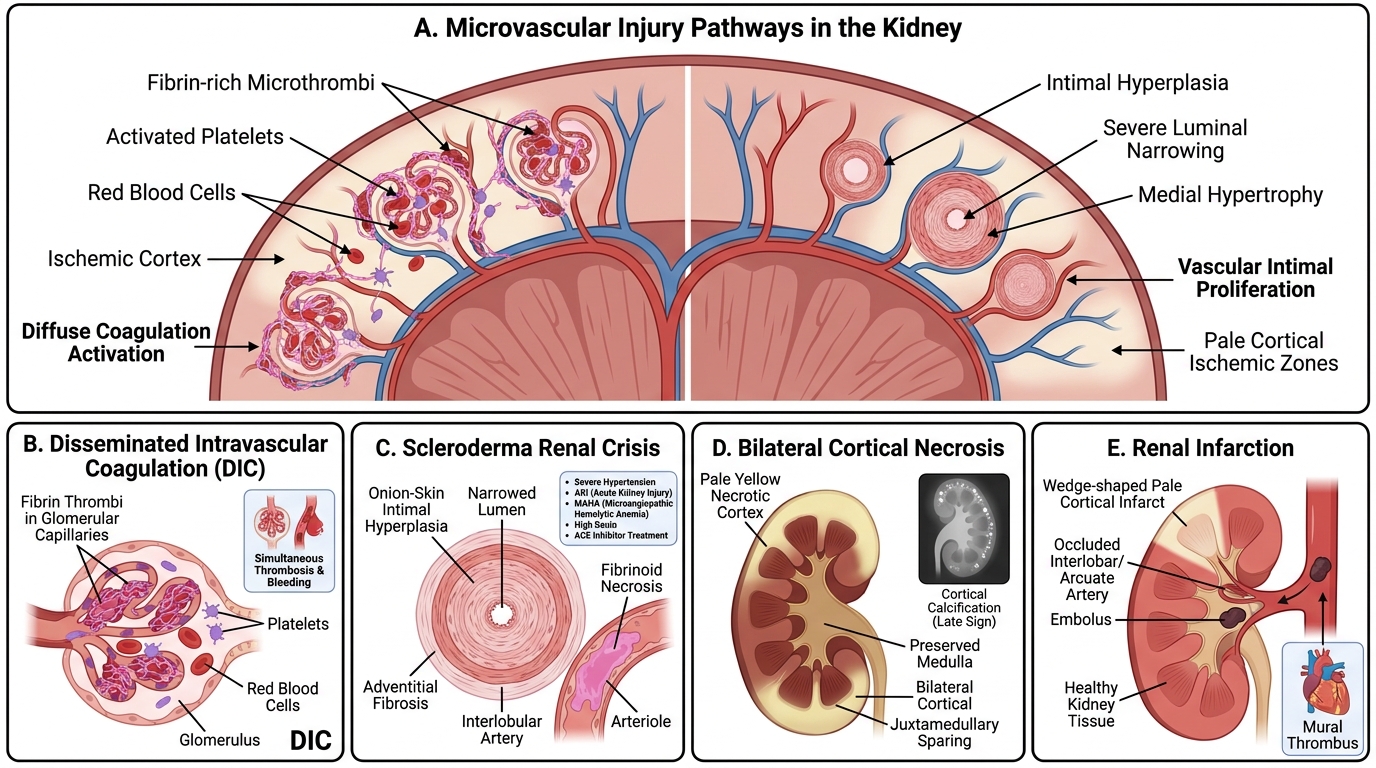
Renal Microvascular Injury: DIC, Scleroderma Crisis, Cortical Necrosis, and Infarction
Disseminated Intravascular Coagulation (DIC):
• Diffuse activation of coagulation and fibrinolysis → microvascular fibrin thrombi + consumption of clotting factors/platelets → simultaneous thrombosis and bleeding
• Renal lesions: fibrin thrombi in glomerular capillaries, cortical ischaemia
• Causes: sepsis (most common), obstetric catastrophes, malignancy, massive transfusion, snake bite
Scleroderma (Systemic Sclerosis) Renal Crisis:
• Sudden severe hypertension + AKI in a scleroderma patient
• Pathogenesis: intimal proliferation + adventitial fibrosis in interlobular arteries → concentric onion-skin hyperplasia (morphologically identical to malignant hypertension) + fibrinoid necrosis of arterioles
• Lab: MAHA picture, elevated plasma renin
• Treatment: ACE inhibitors (captopril) — reduce renin-angiotensin activation; dramatically improved prognosis
Bilateral Cortical Necrosis:
• Ischaemic necrosis of the renal cortex (sparing medulla and a thin juxtamedullary strip)
• Mechanism: prolonged vasospasm + DIC → cortical vessel thrombosis
• Classic obstetric association: abruptio placentae, placenta praevia, eclampsia, septic abortion
• Gross: pale/yellowish cortical zone; medulla preserved (medullary vessels have collateral supply)
• May progress to dystrophic cortical calcification (visible on plain X-ray)
• Outcome: oliguria/anuria → CKD or permanent renal failure
Renal Infarction:
• Usually embolic: atrial fibrillation (left atrial thrombus), infective endocarditis (septic emboli), aortic plaque, post-cardiac catheterisation
• Morphology: wedge-shaped coagulative necrosis oriented cortex-to-papilla, with haemorrhagic rim
• Clinical: sudden flank pain, haematuria, elevated LDH; large infarcts → hypertension (renin release from ischaemic juxtaglomerular cells)
• Treatment: anticoagulation; revascularisation only in acute bilateral occlusion
CLINICAL PEARL
The TMA diagnosis hinges on the peripheral blood film. Whenever you see AKI + low platelets, immediately ask: is there a microangiopathic picture? Order a blood film and look for schistocytes. Schistocytes + thrombocytopenia + AKI = TMA until proven otherwise. Then: (1) check for diarrhoeal prodrome (HUS), (2) look for neurological features (TTP), (3) send ADAMTS13, Shiga-toxin PCR, complement levels. Never transfuse platelets empirically in suspected TTP — it worsens microvascular thrombosis.
SELF-CHECK
Bilateral cortical necrosis is most classically associated with which obstetric condition?
A. Hyperemesis gravidarum in the first trimester
B. Abruptio placentae causing profound hypotension and DIC
C. Premature rupture of membranes at 28 weeks
D. Mild pre-eclampsia with proteinuria in the third trimester
Reveal Answer
Answer: B. Abruptio placentae causing profound hypotension and DIC
Bilateral cortical necrosis classically complicates abruptio placentae and other severe obstetric haemorrhagic/thrombotic catastrophes (eclampsia, septic abortion). The combination of profound circulatory shock and DIC triggers cortical vessel vasospasm and thrombosis, leading to selective cortical ischaemia — the medulla is spared by its collateral supply. Hyperemesis, PROM, and mild pre-eclampsia are not associated with this lesion.
Integrative Summary: Framework for Vascular Renal Disease
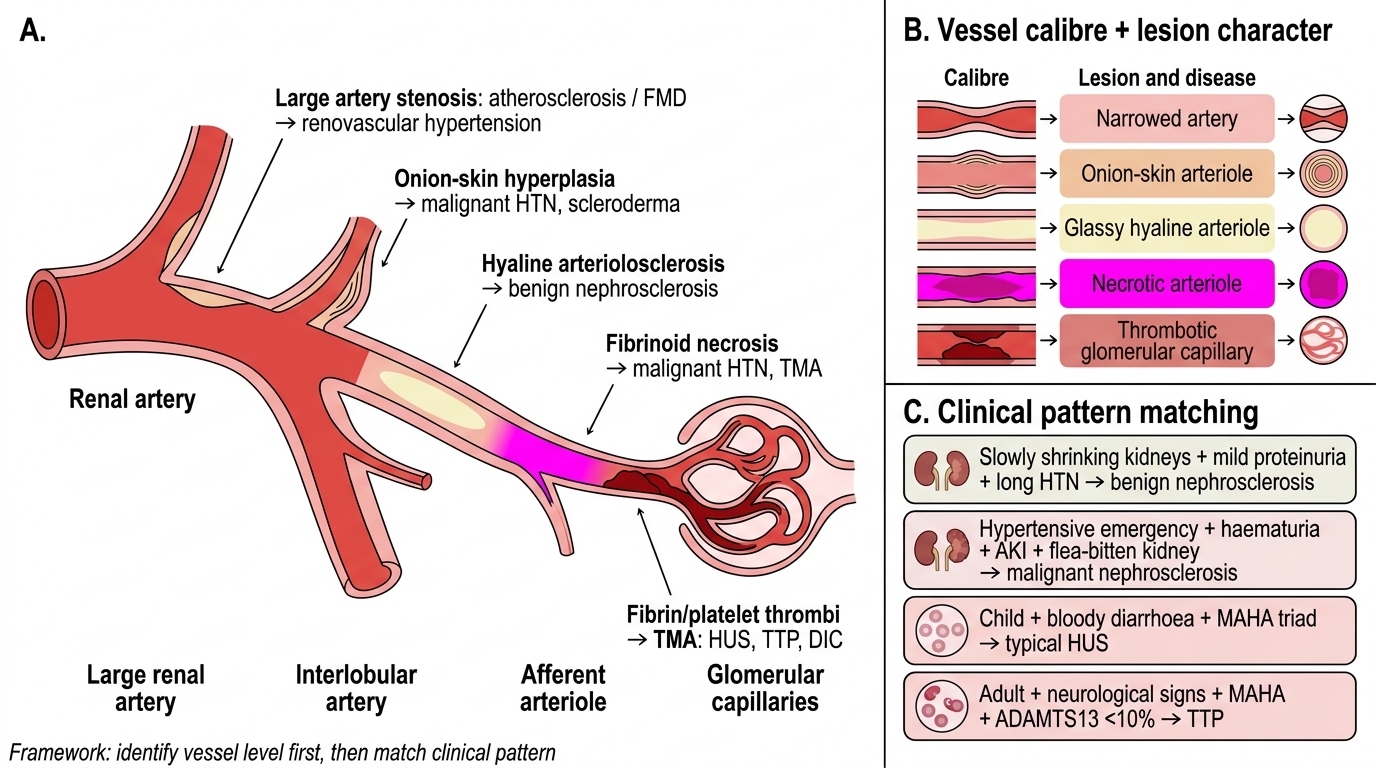
Framework for Vascular Renal Disease
Step 1 — Vessel calibre and lesion character:
| Vessel | Lesion | Disease |
|---|---|---|
| Large artery | Stenosis (atherosclerosis/FMD) | Renovascular hypertension |
| Interlobular artery | Onion-skin hyperplasia | Malignant HTN, scleroderma |
| Afferent arteriole | Hyaline change | Benign nephrosclerosis |
| Afferent arteriole | Fibrinoid necrosis | Malignant HTN, TMA |
| Glomerular capillary | Fibrin/platelet thrombi | TMA (HUS, TTP, DIC) |
Step 2 — Clinical pattern matching:
• Slowly shrinking kidneys + mild proteinuria + long HTN history → Benign nephrosclerosis
• Acute hypertensive emergency + haematuria + AKI + flea-bitten kidney → Malignant nephrosclerosis
• Child + bloody diarrhoea + MAHA triad → Typical HUS (Shiga-toxin; supportive Rx)
• Adult + neurological + MAHA + ADAMTS13 <10% → TTP (plasma exchange is life-saving; no platelets)
• No diarrhoea + relapsing MAHA + complement mutation → aHUS (eculizumab)
• Obstetric catastrophe + DIC + anuria → Cortical necrosis
• Sudden flank pain + AF or endocarditis → Renal infarction
Laboratory approach to suspected TMA:
1. Peripheral blood film (schistocytes?)
2. FBC (Hb, platelets), reticulocyte count, LDH, bilirubin
3. Coombs test (negative in TMA — not immune-mediated haemolysis)
4. Serum creatinine, urinalysis
5. ADAMTS13 activity and inhibitor
6. Stool Shiga-toxin PCR
7. Complement levels (C3, C4, CH50) and CFH mutation screen (if aHUS suspected)