Page 6 of 27
PA27.{11,15} | Vascular Diseases & Thrombotic Microangiopathies — SDL Guide (Part 2)
Renal Artery Stenosis and Renovascular Hypertension
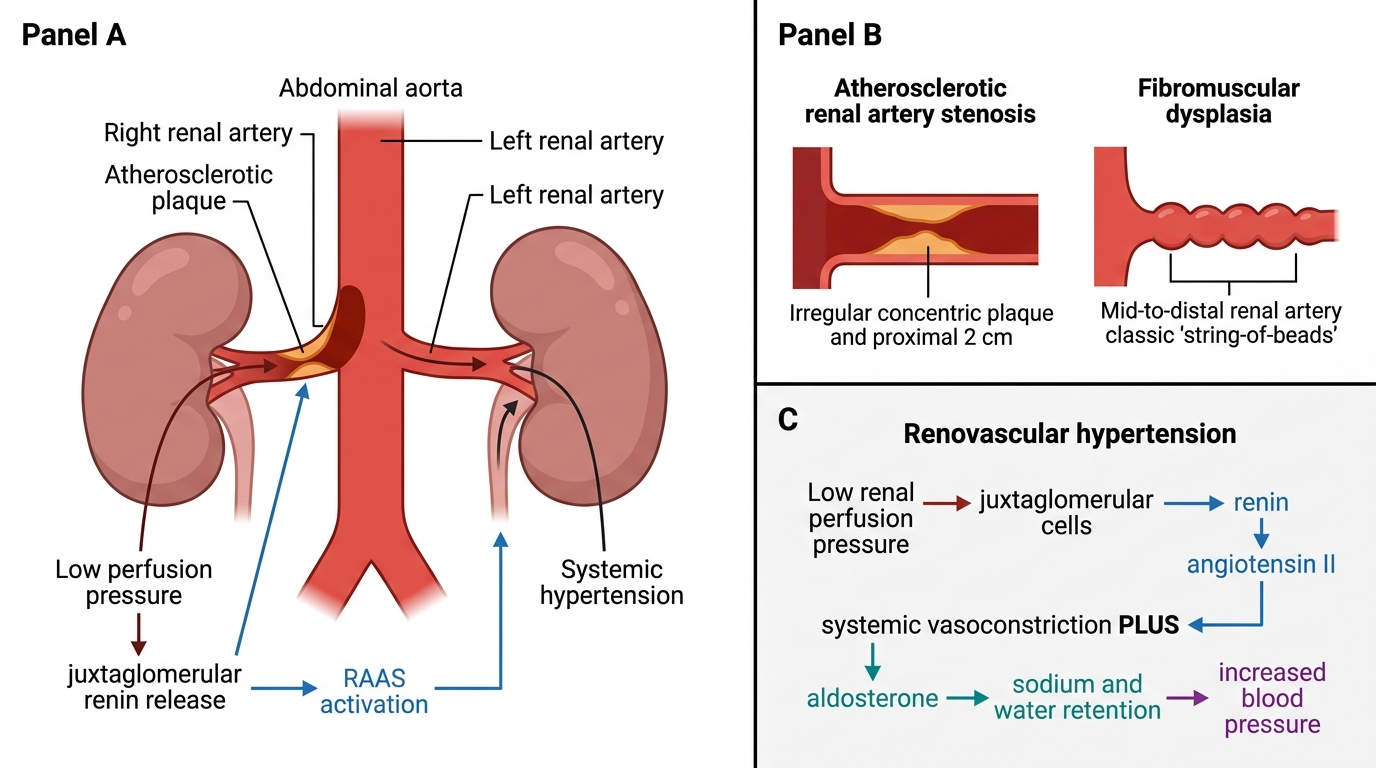
Renal Artery Stenosis and Renovascular Hypertension
Renal artery stenosis (RAS) reduces perfusion pressure to one or both kidneys, activating the RAAS and producing renovascular hypertension — a potentially curable secondary form of hypertension.
Two aetiological types:
| Feature | Atherosclerotic RAS | Fibromuscular dysplasia (FMD) |
|---|---|---|
| Age/sex | Older males, smokers | Young/middle-aged females |
| Location | Proximal (ostium/proximal 2 cm) | Middle and distal renal artery |
| Angiographic appearance | Concentric plaque, irregular stenosis | String-of-beads (alternating stenoses and aneurysms) |
| Laterality | Usually unilateral | Usually unilateral; bilateral in 30% |
Pathogenesis of hypertension:
Reduced renal perfusion → juxtaglomerular cells sense low pressure → renin release → Ang II → systemic vasoconstriction + aldosterone → BP rises. The contralateral normal kidney is suppressed — exposed to high BP but normal perfusion-pressure RAAS dynamics.
Renal morphology:
• Ipsilateral kidney: ischaemic nephropathy — atrophic, small, with tubular atrophy, interstitial fibrosis, and juxtaglomerular cell hyperplasia
• If untreated → permanent renal atrophy
Diagnosis: captopril-enhanced isotope renogram, Doppler ultrasound, CT/MR angiography.
Treatment: angioplasty ± stent (FMD responds excellently); ACE inhibitor is CONTRAINDICATED in bilateral stenosis → causes acute renal failure by removing the efferent arteriolar tone that maintains GFR.
Thrombotic Microangiopathy: Unifying Concept
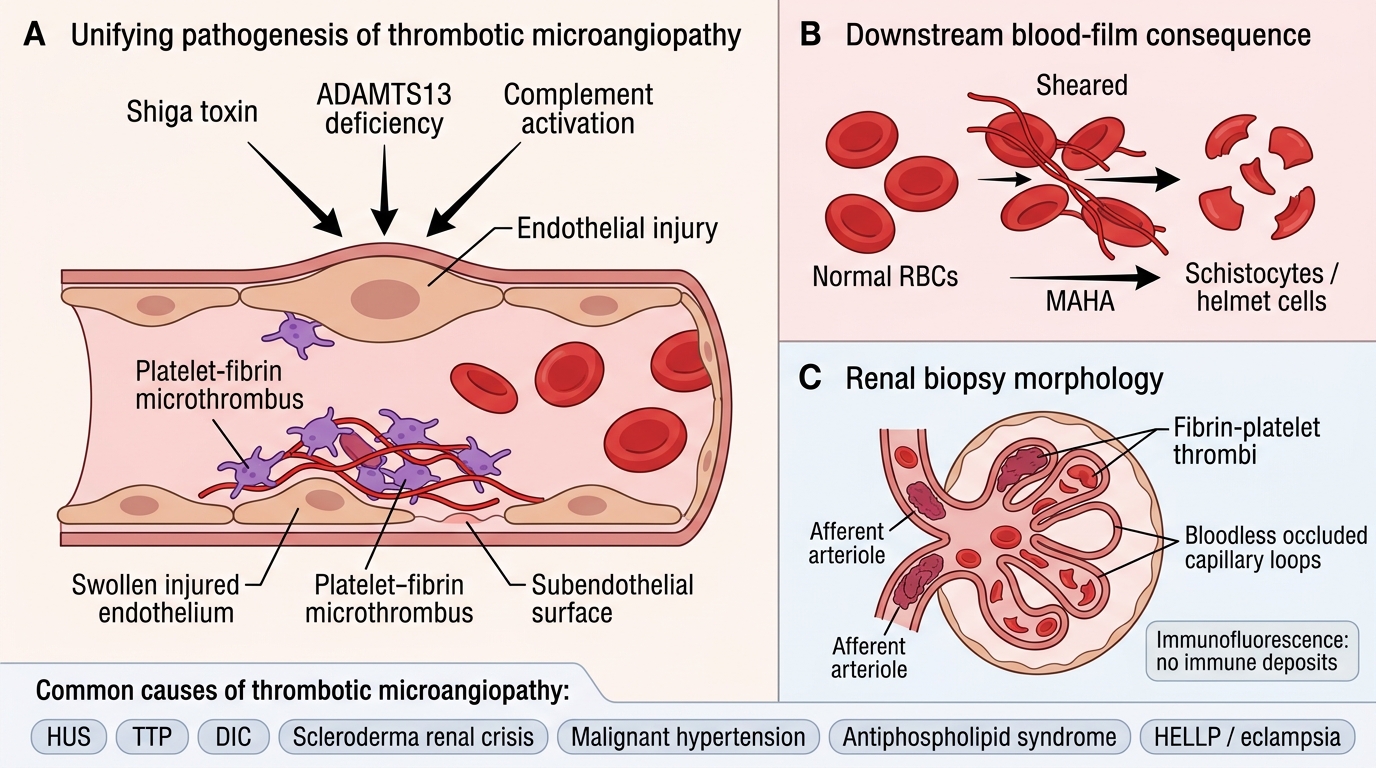
Thrombotic Microangiopathy: Unifying Mechanism
Thrombotic microangiopathy (TMA) is not a single disease but a pathological syndrome defined by:
1. Endothelial injury in small vessels (arterioles and capillaries)
2. Formation of platelet-fibrin microthrombi in vessel lumina
3. Downstream consequences:
- Microangiopathic haemolytic anaemia (MAHA) — RBCs sheared by fibrin strands → schistocytes (helmet cells, fragmented RBCs) on blood film
- Thrombocytopenia — platelets consumed in thrombi
- Acute kidney injury (AKI) — glomerular and arteriolar microthrombi → ischaemia
Shared renal morphology (regardless of cause):
• Fibrin/platelet thrombi in glomerular capillaries, afferent arterioles, interlobular arteries
• Glomerular capillary wall thickening (bloodless glomeruli — capillaries occluded, no blood)
• Schistocytes and fragmented RBCs visible in glomerular capillaries on biopsy
• No immune deposits on immunofluorescence (distinguishes TMA from immune-mediated GN)
TMA causes — the unifying mechanism operates in:
• HUS (typical and atypical)
• TTP
• DIC
• Scleroderma renal crisis
• Malignant hypertension (overlap)
• Antiphospholipid syndrome
• Pregnancy-associated (HELLP, eclampsia)
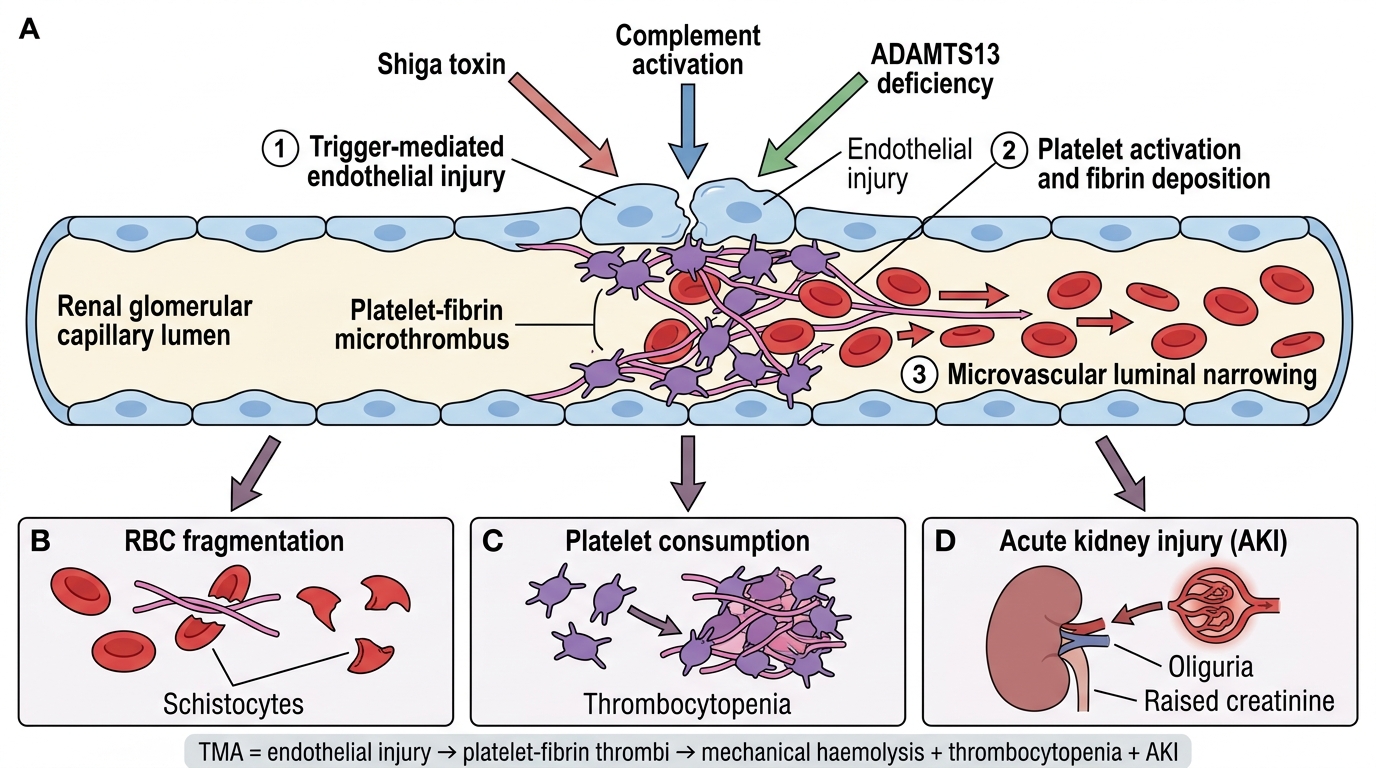
Mechanism of Thrombotic Microangiopathy
SELF-CHECK
A 7-year-old presents with bloody diarrhoea followed by oliguria, pallor, and bruising. Blood film shows fragmented red blood cells. Platelets 22,000/μL, haemoglobin 7.2 g/dL, creatinine elevated. What is the mechanism of anaemia in this condition?
A. Autoimmune IgG against red cell antigens (warm AIHA)
B. Bone marrow suppression by bacterial exotoxin
C. Mechanical fragmentation of RBCs by intravascular fibrin strands
D. Direct complement-mediated lysis of red blood cells
Reveal Answer
Answer: C. Mechanical fragmentation of RBCs by intravascular fibrin strands
This is classic HUS (typical/childhood form) following Shiga-toxin-producing E. coli O157:H7. The anaemia is microangiopathic — fibrin strands deposited in glomerular capillaries shear passing RBCs into schistocytes (fragmented cells). This is mechanical intravascular haemolysis: Coombs-negative (not immune), no complement-mediated lysis (as in PNH). Bone marrow suppression does not produce schistocytes on blood film.
Haemolytic Uraemic Syndrome (HUS)
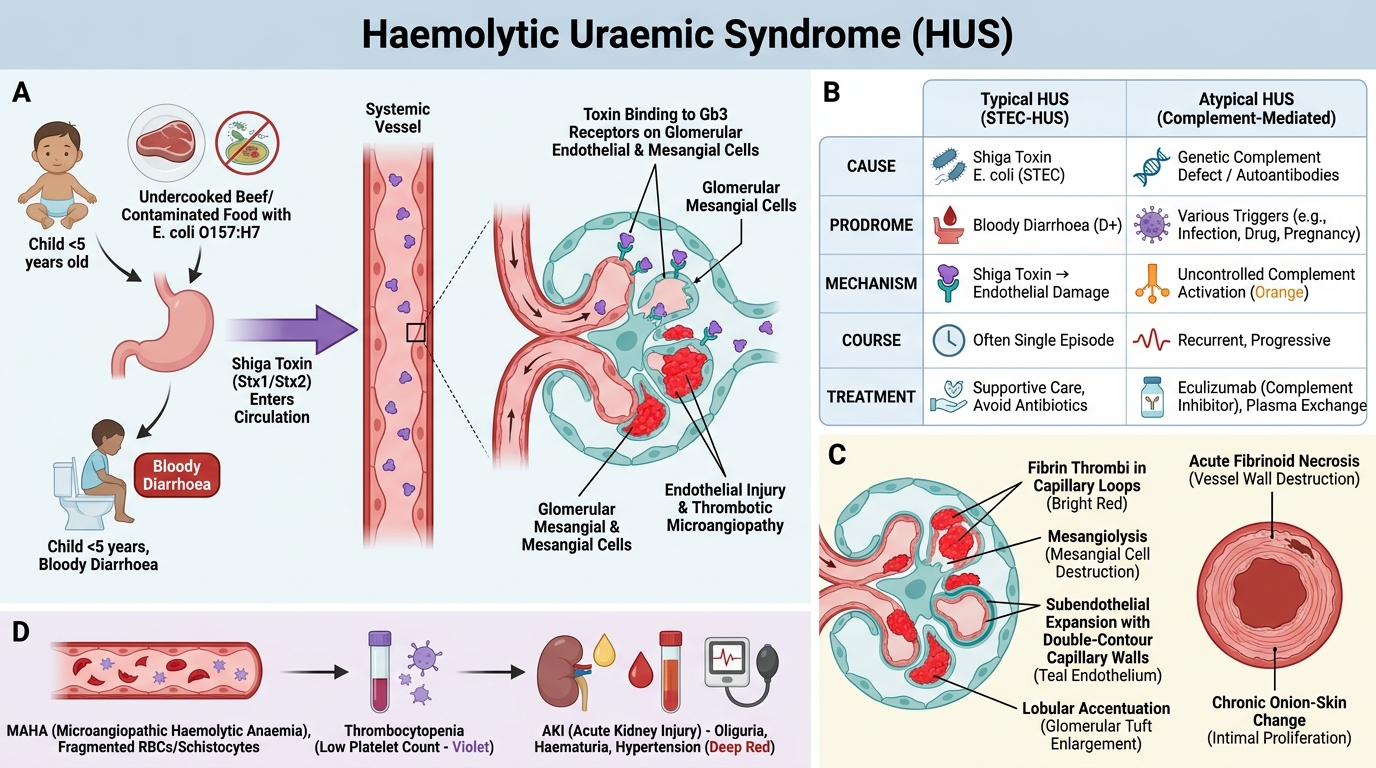
Haemolytic Uraemic Syndrome: Pathogenesis, Types and Renal Morphology
Typical (Childhood / D+ HUS):
- Cause: Shiga toxin (Stx1 or Stx2) produced by E. coli O157:H7 (haemorrhagic colitis, undercooked beef) or Shigella dysenteriae type 1
- Epidemiology: Children under 5; preceded by bloody diarrhoea (D+HUS = diarrhoea-associated)
- Pathogenesis: Stx absorbed from gut → systemic circulation → binds Gb3 receptor on glomerular endothelial cells and mesangial cells (Gb3 highly expressed in child kidneys — explains age predilection) → endothelial activation/injury → TMA
- Clinical triad: MAHA + thrombocytopenia + AKI → oliguria, haematuria, hypertension
- Prognosis: ~90% recover with supportive care; 5-10% progress to CKD
Atypical (aHUS / D- HUS):
- Cause: Complement pathway dysregulation — mutations in complement factor H (CFH), factor I, membrane cofactor protein (MCP/CD46), or gain-of-function C3/CFB mutations; also anti-CFH antibodies
- Mechanism: Uncontrolled alternative complement activation → endothelial injury → TMA (no diarrhoeal prodrome)
- Course: More severe, relapsing; 50% progress to ESRD
- Treatment: Eculizumab (anti-C5 monoclonal antibody — blocks terminal complement activation) — transformative therapy for aHUS
Key renal morphology (both HUS types):
• Glomerular: fibrin thrombi, mesangiolysis, capillary wall double-contour (subendothelial expansion), lobular pattern
• Arteriolar: fibrinoid necrosis (acute); onion-skin changes (chronic recurrent)
• Schistocytes visible in capillary lumina on biopsy