Page 1 of 16
PA33.1-3 | Skin Cancers: BCC, SCC & Melanoma — SDL Guide
Learning Objectives
- Describe the risk factors, pathogenesis, gross/histological pathology, and natural history of squamous cell carcinoma (SCC) of the skin, including its premalignant precursor actinic keratosis (PA33.1)
- Describe the risk factors, pathogenesis, gross/histological pathology, and natural history of basal cell carcinoma (BCC) of the skin, explaining why it is locally destructive but almost never metastasises (PA33.2)
- Distinguish a benign melanocytic naevus from melanoma using the ABCDE criteria, and describe the aetiology, pathogenesis (BRAF/NRAS), radial vs vertical growth phases, Breslow depth, histological subtypes, immunohistochemical markers, and metastatic behaviour of melanoma (PA33.3)
INSTRUCTIONS
You are standing at the crossroads of cell biology and clinical oncology. Skin cancers are the most common cancers worldwide, and India's UV burden, fair-skinned populations, and increasing immunosuppression rates are making them a growing clinical challenge. BCC, SCC, and melanoma are distinct tumours with different cell origins, mutation profiles, behaviours, and prognoses — and a pathologist's ability to distinguish them on a glass slide can be the difference between a simple excision and a life-saving immunotherapy referral. Work through this SDL in order: normal skin anchor → premalignant precursors → keratinocyte cancers (SCC then BCC) → melanocytic spectrum (naevus → melanoma). Estimated reading time: 35–40 minutes.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch. 25 — The Skin (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 58-year-old farmer from Rajasthan comes to your OPD with a pearly, slow-growing nodule on his nose. He has had it for two years and ignored it because it 'didn't hurt.' His neighbour, also a farmer, has a red, scaly plaque on his forearm that recently started ulcerating and bleeding. And last week, a 45-year-old software engineer visited with a mole on his back that his wife noticed had grown and changed colour over six months.
Three patients, three different lesions — but all three come down to the same question: which cells went wrong, how, and what happens next? Skin cancer is not one disease. Understanding the three major forms — BCC, SCC, and melanoma — requires you to link cell biology to clinical behaviour. Let's do exactly that.
WHY THIS MATTERS
Skin cancers are the most common cancers in humans globally. In India, while the incidence is lower than in Australia or the US due to higher melanin content in most of the population, the numbers are rising. More importantly for your clinical practice:
- You will diagnose skin lesions at the bedside before histology is available — the ABCDE criteria you learn today are a real clinical tool
- BCC is so common that a general physician or dermatologist may see one every week
- SCC arising in chronic wounds, burns, and sinuses is disproportionately common in India
- Melanoma, though rarer, carries the highest mortality of the three and presents diagnostic challenges you must not miss
- Immunosuppressed patients (transplant recipients, HIV patients) have dramatically elevated risk for all three — increasingly relevant in urban India
PA33.1, PA33.2, and PA33.3 are NMC 2024 core competencies that will appear in your professional examinations and, more importantly, in your clinical practice from internship onwards.
RECALL
Before we dive in, activate what you already know:
- Epidermis layers (from deep to surface): stratum basale → stratum spinosum → stratum granulosum → stratum lucidum (only in thick skin) → stratum corneum. The basal layer contains keratinocyte stem cells and melanocytes (1 melanocyte : 10 keratinocytes).
- Keratinocytes produce keratin (a structural protein) and differentiate upwards — each layer is a step in the cornification programme.
- UV radiation causes DNA damage, primarily cyclobutane pyrimidine dimers at C–C, T–T, and C–T junctions. The tumour suppressor TP53 normally detects this damage and triggers either repair or apoptosis.
- Melanin is made by melanocytes from tyrosine via tyrosinase and transferred to keratinocytes via melanosomes — it absorbs UV radiation and protects the nucleus.
- From your Pathology of General Principles module: carcinogenesis requires initiating mutations + promotional stimuli; oncogenes accelerate growth, tumour suppressor genes (like TP53, PTCH) brake it.
With this foundation, the pathogenesis of all three skin cancers will make mechanistic sense.
Normal Skin and the UV Damage Threshold
Skin architecture is the essential starting point. The skin has two main layers:
- Epidermis — stratified squamous epithelium; avascular; keratinocytes (90%), melanocytes (8%), Langerhans cells (2%), Merkel cells (<1%)
- Dermis — collagen-rich connective tissue; contains blood vessels, nerves, hair follicles, sweat glands, sebaceous glands
The dermo-epidermal junction (DEJ) is a basement membrane zone containing type IV collagen and laminin. Invasion through the DEJ defines malignancy in skin epithelial tumours — this distinction is critical for distinguishing in-situ from invasive SCC.
In terms of UV damage:
- UVB (280–315 nm) — directly absorbed by DNA → pyrimidine dimers → TP53 mutation (the 'guardian of the genome'). This is the dominant carcinogen in SCC and BCC.
- UVA (315–400 nm) — penetrates deeper; generates reactive oxygen species (ROS); damages melanocytes; implicated in melanoma pathogenesis.
- Melanin provides photoprotection — people with Fitzpatrick skin types I–II (lighter skin) are at highest risk for all UV-driven skin cancers.
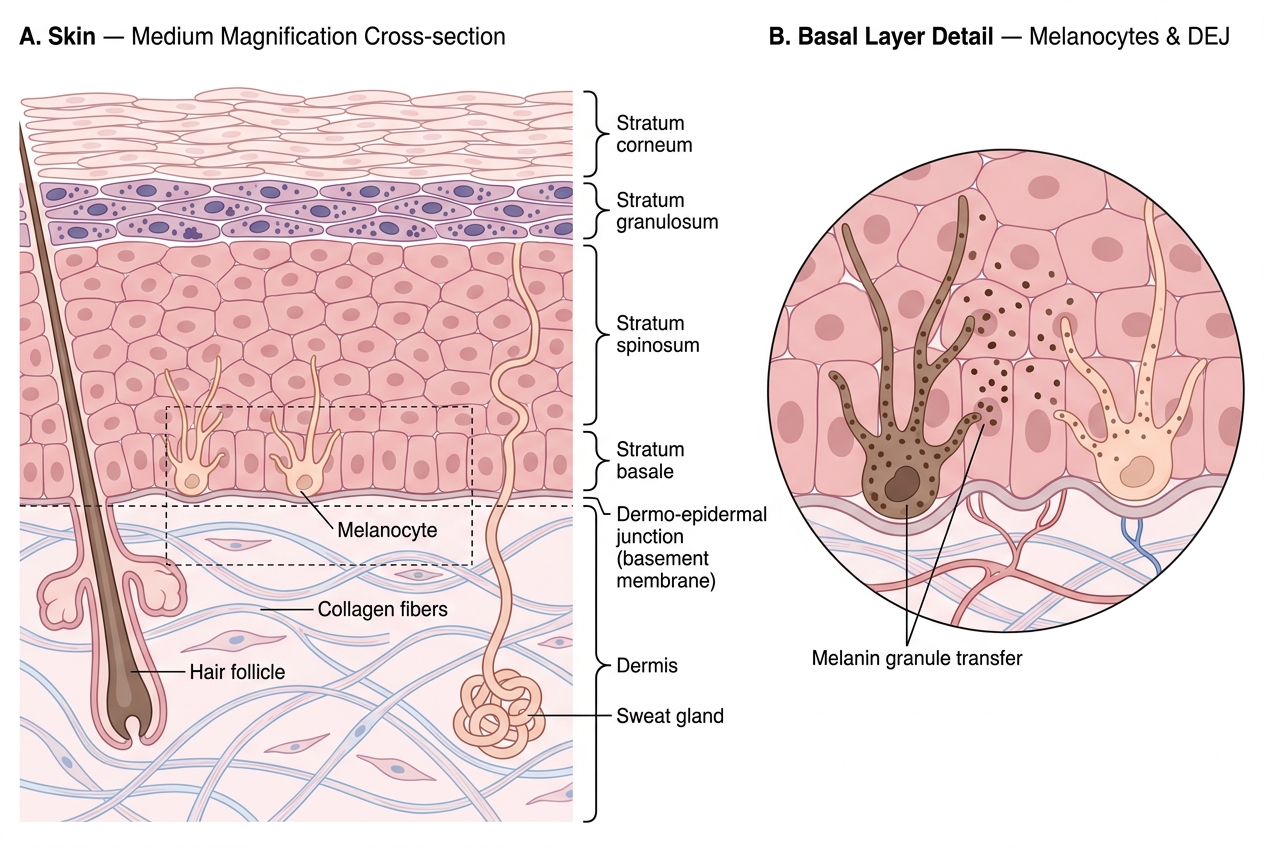
Skin Histology — Epidermal Strata, Melanocytes, and Dermo-Epidermal Junction
The concept of the UV damage threshold: repeated sub-lethal UV doses accumulate somatic mutations in keratinocytes and melanocytes over years to decades. This explains why skin cancers are predominantly tumours of sun-exposed areas in older adults — a field effect where the entire exposed skin surface accumulates mutations.
Actinic (Solar) Keratosis — The Premalignant Warning Sign
Actinic keratosis (AK) — also called solar keratosis — is the most important premalignant lesion of the skin. It is the direct precursor of invasive SCC.
Epidemiology and risk factors:
• Chronic, cumulative UV (sun) exposure — outdoor workers, farmers, gardeners
• Fitzpatrick types I–II (fair skin, burns easily)
• Age >50 years (decades of cumulative UV)
• Immunosuppression (transplant patients develop AKs in their 30s–40s)
Gross/clinical appearance:
• Rough, scaly, erythematous (red) plaques on sun-exposed skin: face, scalp (bald men), dorsum of hands, forearms
• Sandpaper texture felt before it is seen — the key clinical finding
• Usually <1 cm; multiple lesions common (field cancerisation)
Histology:
• Atypical keratinocytes confined to the lower layers of the epidermis (basal and spinous)
• Dyskeratosis (premature, abnormal cornification of individual cells)
• Solar elastosis in the dermis — degenerated elastic fibres appear basophilic (blue) rather than the normal pink eosinophilic collagen; this is direct evidence of UV damage in the dermis
• NO breach of the basement membrane — this is in-situ disease
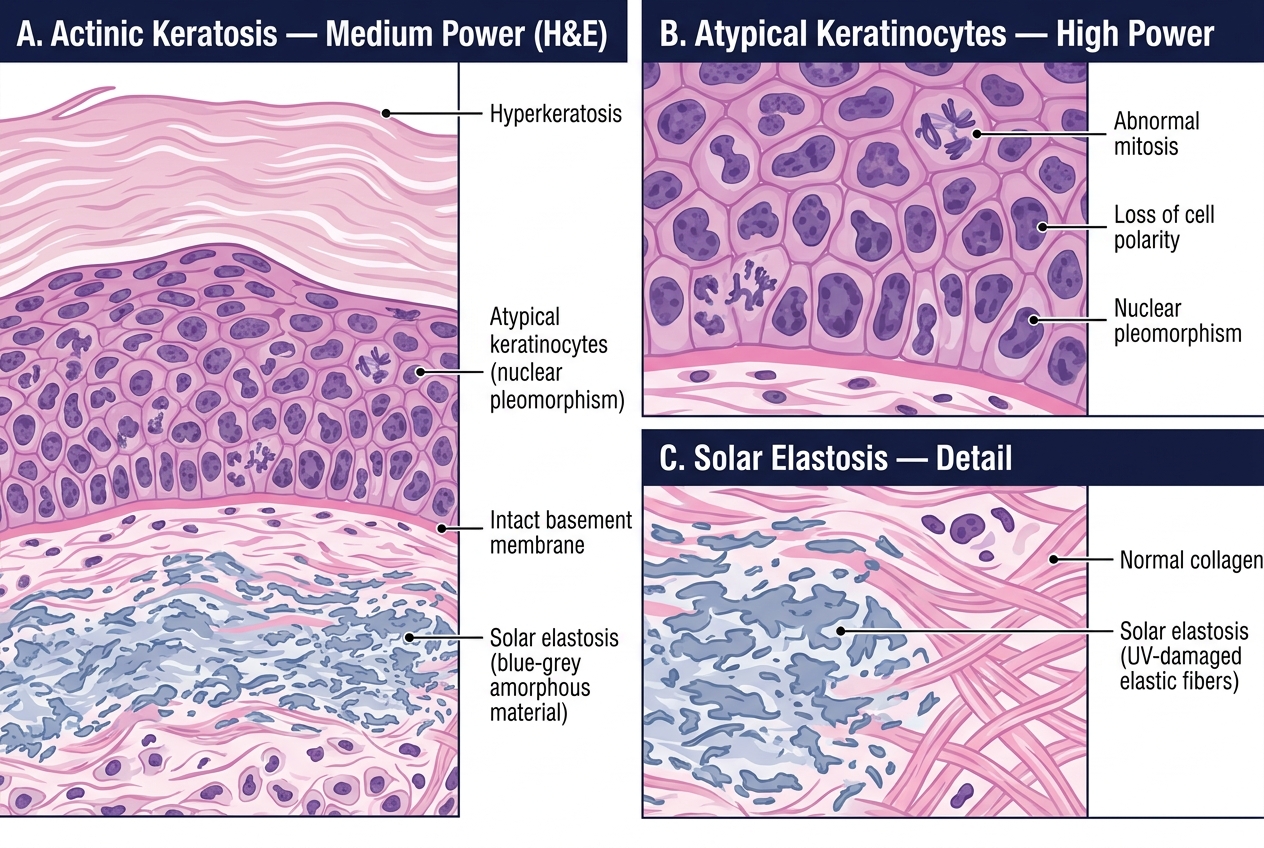
Histology of Actinic Keratosis — Medium and High Power
Natural history: ~0.1–1% of AKs per year progress to invasive SCC. However, patients with multiple AKs have cumulative risk. Regression occurs in up to 25% untreated. Treatment (topical 5-FU, imiquimod, cryotherapy, photodynamic therapy) aims to prevent this progression.
Clinical pearl: The term "field cancerisation" means that when you see one AK, the surrounding skin has the same UV damage. Treating the visible lesion without treating the field leaves the patient at risk — this is why topical treatments covering the entire field are preferred over spot-treating alone.
Squamous Cell Carcinoma (SCC) — Pathogenesis and Pathology (PA33.1)
Squamous cell carcinoma is a malignant tumour of keratinocytes (the predominant cells of the epidermis). It is the second most common skin cancer after BCC.
Risk factors:
| Risk Factor | Mechanism |
|---|---|
| Chronic UV/sun exposure | TP53 mutation, DNA pyrimidine dimers |
| Immunosuppression (transplant, HIV, steroids) | Loss of immune surveillance — 65× increased risk post-transplant |
| HPV (types 16, 18) | E6 protein degrades TP53; E7 inhibits Rb |
| Chronic inflammation/scarring | Marjolin's ulcer (SCC in chronic wounds, burn scars, sinus tracts) |
| Chemical carcinogens | Arsenic (Bowen's disease), polycyclic aromatic hydrocarbons (chimney sweep) |
| Ionising radiation | X-ray therapy sites |
| Precursor lesions | Actinic keratosis, Bowen's disease, leukoplakia (oral), erythroplasia of Queyrat (penis) |
Pathogenesis:
The central molecular event is TP53 mutation — induced by UV-generated pyrimidine dimers. TP53 normally triggers either DNA repair (via p21-mediated cell cycle arrest) or apoptosis. Mutant TP53 is non-functional → damaged keratinocytes survive and proliferate instead of dying → accumulation of further mutations → full malignant transformation.
Additional pathways: CDKN2A loss (p16 inactivation → uncontrolled Rb phosphorylation → unchecked cell cycle entry), Ras activation.
Bowen's disease — SCC in situ:
Bowen's disease is full-thickness epidermal atypia without basement membrane breach — i.e., SCC in situ.
Histology:
• Atypical keratinocytes throughout the full thickness of the epidermis (unlike AK, where atypia is confined to the lower layers)
• Windblown appearance — cells lose their normal polarity; nuclei are large, irregular, hyperchromatic
• Normal basket-weave stratum corneum is replaced by parakeratosis (nuclei retained in the cornified layer)
• Intact basement membrane — no invasion
• Individual cell dyskeratosis
Invasive SCC — histology:
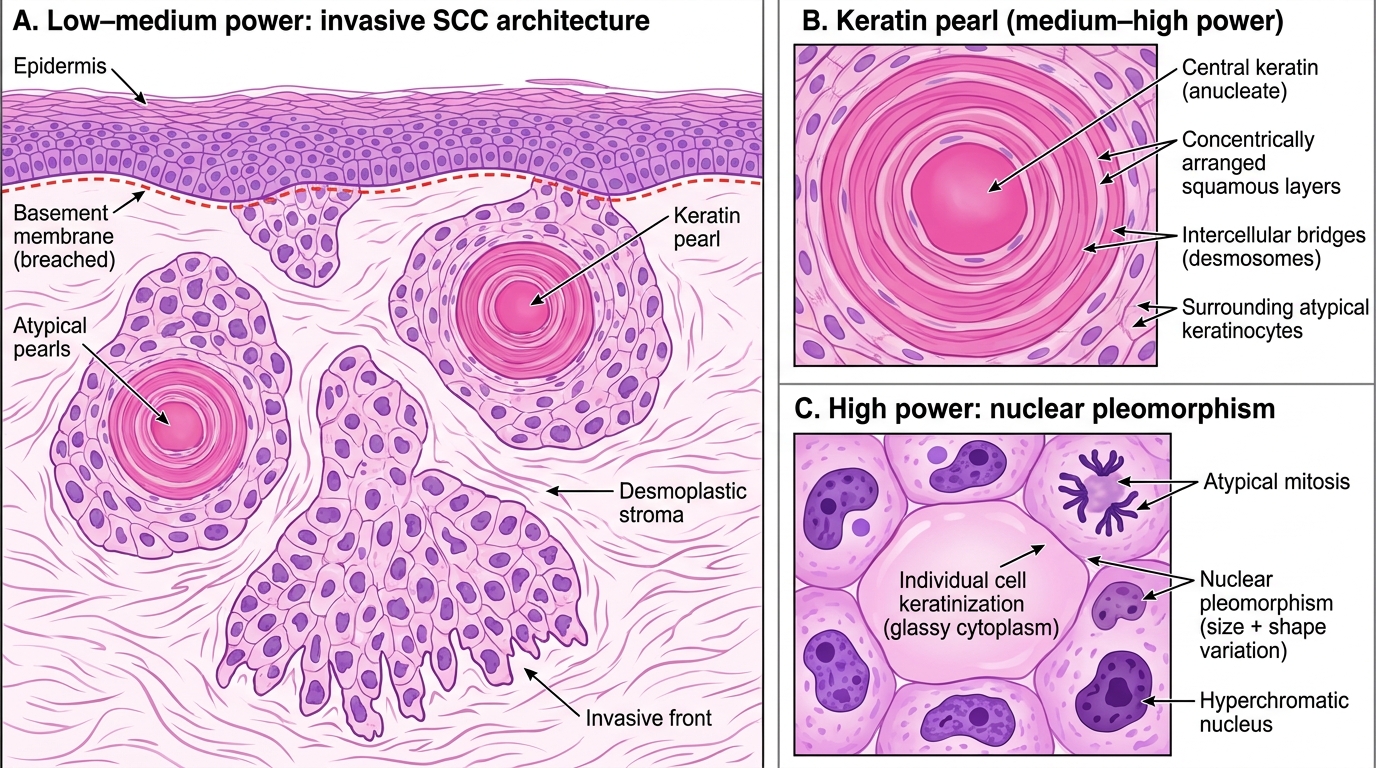
Histology of Invasive Squamous Cell Carcinoma (H&E)
Key histological features of invasive SCC:
• Keratin pearls (also called epithelial/squamous pearls) — concentric whorls of squamous cells undergoing premature cornification; pathognomonic of squamous differentiation; seen in well-differentiated SCC
• Individual cell keratinisation — large, pink, glassy cytoplasm in atypical cells
• Intercellular bridges (desmosomes) visible on H&E in well-differentiated tumours
• Invasion into dermis and deeper — through the basement membrane
• Desmoplastic stroma — reactive fibrous stroma surrounding tumour nests
• Tumour grading: Well-differentiated (keratin pearls present) → Moderately differentiated → Poorly differentiated (no pearls, little evidence of squamous origin)
Gross appearance:
• Indurated (firm), elevated, ulcerated plaque or nodule on sun-exposed skin
• Central ulceration with rolled or everted edges
• May arise in a pre-existing AK, scar, or chronic ulcer
Natural history:
SCC of the skin has a low but real metastatic potential — this distinguishes it from BCC.
- Overall metastatic rate: 2–5% for typical sun-induced SCC
- High-risk features for metastasis: size >2 cm, depth >4 mm, poor differentiation, perineural invasion, immunosuppression, Marjolin's ulcer, SCC at specific sites (lip, ear)
- Metastatic route: lymph node first → regional nodes → distant (lung, liver, bone)
- Marjolin's ulcer (SCC arising in chronic ulcers, burn scars, osteomyelitis sinuses) has much higher metastatic rate (~30%) — possibly because chronic inflammation drives more aggressive phenotypes
5-year cure rate for localised SCC with excision: >90%. Metastatic disease: poor prognosis.
CLINICAL PEARL
Marjolin's ulcer is a squamous cell carcinoma that arises in a chronically inflamed, scarred, or ulcerated site — classically a chronic venous ulcer, burn scar, or osteomyelitis sinus. It is characterised by a long latent period (often 20–40 years) between the initial injury and malignant transformation. The diagnosis is frequently missed because the new growth is attributed to the existing wound. Key clinical clues: a chronic wound that suddenly changes in character, develops a raised/indurated edge, bleeds easily, or fails to heal with standard treatment. Histology confirms invasive SCC. Marjolin's ulcers have a significantly higher metastatic rate (~30%) compared to sun-induced SCC because the inflammatory milieu drives more aggressive phenotypes and the tumour may be deep by the time it is diagnosed.