Page 11 of 24
PA2.{3,6-7} | Cellular Adaptations, Accumulations & Aging — SDL Guide (Part 3)
Intracellular Accumulations — Lipids
When cells are stressed or injured, they may accumulate abnormal amounts of normal substances, or store abnormal substances. The main categories are lipids, proteins, glycogen, and pigments.
Lipid accumulations:
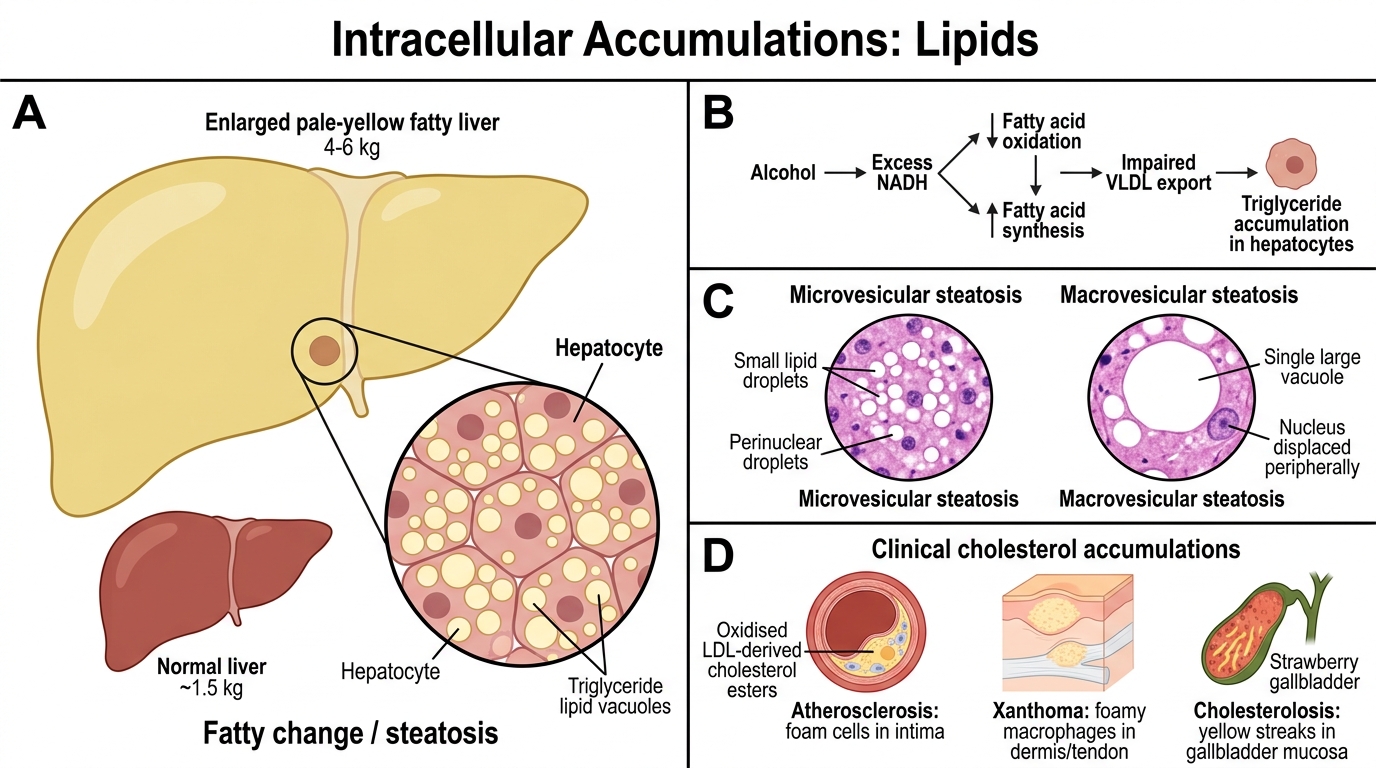
Fatty change (steatosis): Accumulation of triglycerides within parenchymal cells, most commonly hepatocytes. The liver is most vulnerable because it is the main site of lipid processing.
Causes in liver: alcohol (most common in high-income countries), obesity/metabolic syndrome (NAFLD/NASH), diabetes mellitus, protein malnutrition (kwashiorkor), hepatotoxins (CCl₄), pregnancy.
Mechanism in alcoholic fatty liver: Alcohol → excess NADH → ↓ fatty acid oxidation, ↑ fatty acid synthesis, impaired VLDL export → triglycerides accumulate in hepatocytes.
Morphology: Enlarged pale-yellow liver (up to 4–6 kg, normal ~1.5 kg). H&E: clear cytoplasmic vacuoles (lipid dissolved during processing) — small droplets initially perinuclear (microvesicular steatosis, e.g., acute fatty liver of pregnancy, Reye syndrome), then large single vacuoles displacing the nucleus (macrovesicular steatosis, alcohol/NAFLD).
Cholesterol accumulations:
• Atherosclerosis — foam cells in intima are macrophages engorged with oxidised LDL-derived cholesterol esters
• Xanthomas — clusters of foamy macrophages in dermis/tendons in hypercholesterolaemia
• Cholesterolosis — gallbladder mucosa yellow streaks ('strawberry gallbladder')
Intracellular Lipid Accumulations
Intracellular Accumulations — Proteins, Glycogen & Pigments
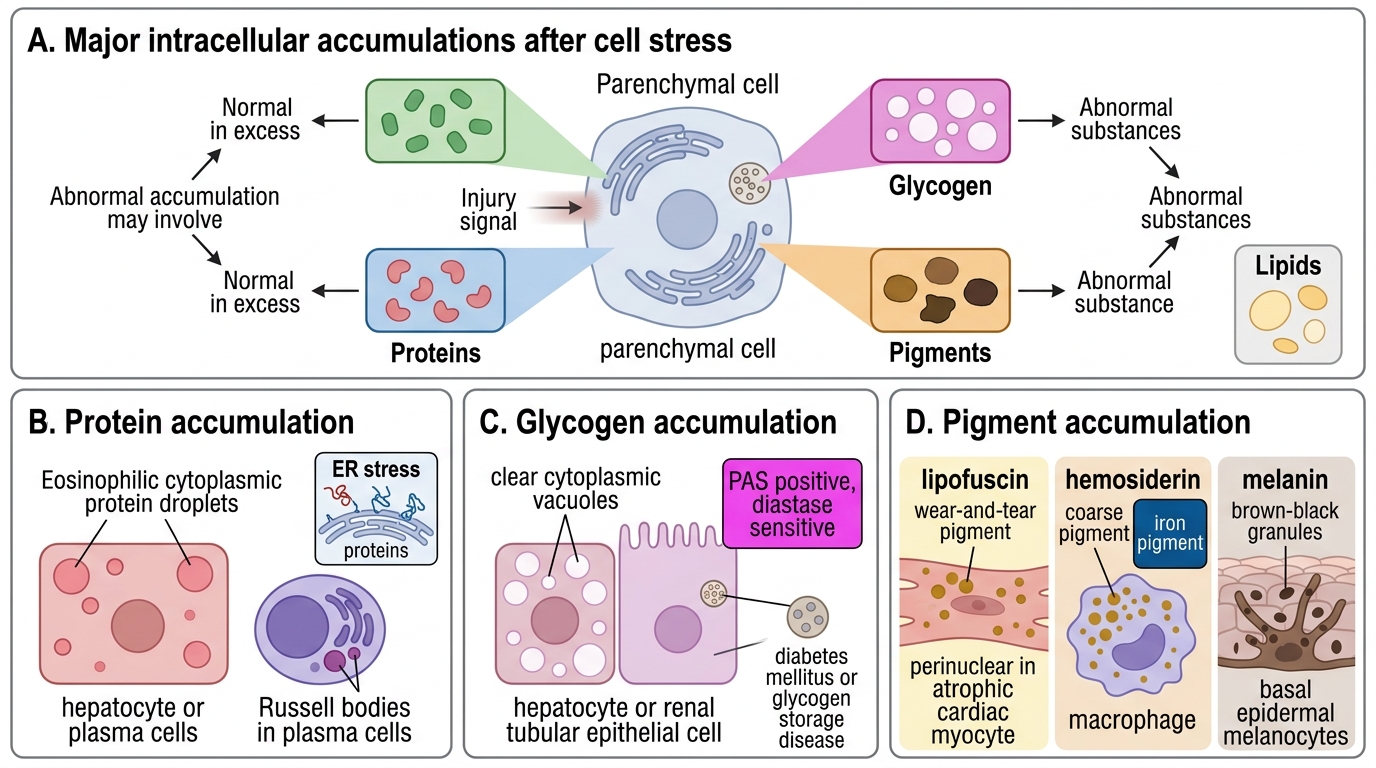
Protein accumulations:
• Mallory–Denk bodies (Mallory's hyaline): Eosinophilic cytoplasmic inclusions in hepatocytes of alcoholic hepatitis; composed of aggregated intermediate filaments (keratins 8 and 18). A hallmark of alcoholic liver disease but also seen in NASH, Wilson's disease.
• Russell bodies: Distended ER of plasma cells packed with immunoglobulin that cannot be secreted; eosinophilic, grape-like intracytoplasmic inclusions.
• Protein droplets in proximal tubular epithelium (nephrotic syndrome — reabsorbed albumin).
Glycogen accumulations:
• Normally present in liver and skeletal muscle as an energy store.
• Excess glycogen: glycogen storage diseases (e.g., von Gierke, Pompe) — enzyme defects preventing glycogen breakdown.
• In diabetic patients — glycogen accumulates in tubular epithelium, loop of Henle, and hepatocyte nuclei ('glycogenic hepatopathy').
• Morphology: clear, non-lipid vacuoles that stain with PAS (periodic acid-Schiff) and are PAS-diastase digestible.
Pigment accumulations:
Exogenous pigments:
• Anthracosis (carbon pigment): Most common exogenous pigment. Inhaled carbon particles are engulfed by alveolar macrophages → travel to hilar lymph nodes and lung interstitium → black pigmentation. Ubiquitous in urban dwellers, smokers. Associated with progressive massive fibrosis in coal miners.
Endogenous pigments:
• Lipofuscin ('wear-and-tear' pigment): Insoluble yellowish-brown granules; complexes of lipid + protein generated by peroxidation of cellular membranes during autophagy. Accumulates in non-dividing cells (neurons, cardiac myocytes, liver) with ageing. Marker of cellular senescence; does not itself cause injury ('benign' pigment).
• Melanin: Brown-black pigment produced by melanocytes via tyrosinase; stored in melanosomes. Increased in Addison's disease, melanoma; decreased/absent in albinism.
• Haemosiderin: Golden-yellow, coarse granular pigment; derived from ferritin micelles when iron storage exceeds transferrin-binding capacity. Stains blue with Perl's Prussian blue reaction. Localised haemosiderin: old haematoma ('bruise turns yellow-brown'), pulmonary haemosiderosis (heart failure cells). Systemic haemosiderosis: widespread haemosiderin in multiple organs (liver, spleen, bone marrow, skin) without tissue damage — from haemolysis, multiple transfusions, dietary iron overload.
• Bilirubin: Yellow-green pigment (conjugated bilirubin in bile ducts, unconjugated in hepatocytes in severe jaundice); bile plugs in bile canaliculi visible on H&E in cholestasis.
Intracellular Accumulations: Proteins, Glycogen and Pigments
SELF-CHECK
On autopsy of a 68-year-old who died of congestive heart failure, the lungs contain macrophages filled with golden-brown granules that stain positively with Perl's Prussian blue reaction. What is the most likely identity of this pigment?
A. Haemosiderin
B. Lipofuscin
C. Carbon (anthracosis)
D. Bilirubin
Reveal Answer
Answer: A. Haemosiderin
Haemosiderin stains blue with Perl's Prussian blue reaction (the iron-specific stain). In chronic left heart failure, elevated pulmonary venous pressure causes repeated microhaemorrhages into the alveoli; alveolar macrophages phagocytose red cells and degrade haemoglobin to haemosiderin — these are called 'heart failure cells'. Lipofuscin is yellow-brown and Perl's negative. Carbon pigment is black. Bilirubin is yellow-green.
Cellular Aging — Mechanisms
Mechanisms of Cellular Aging
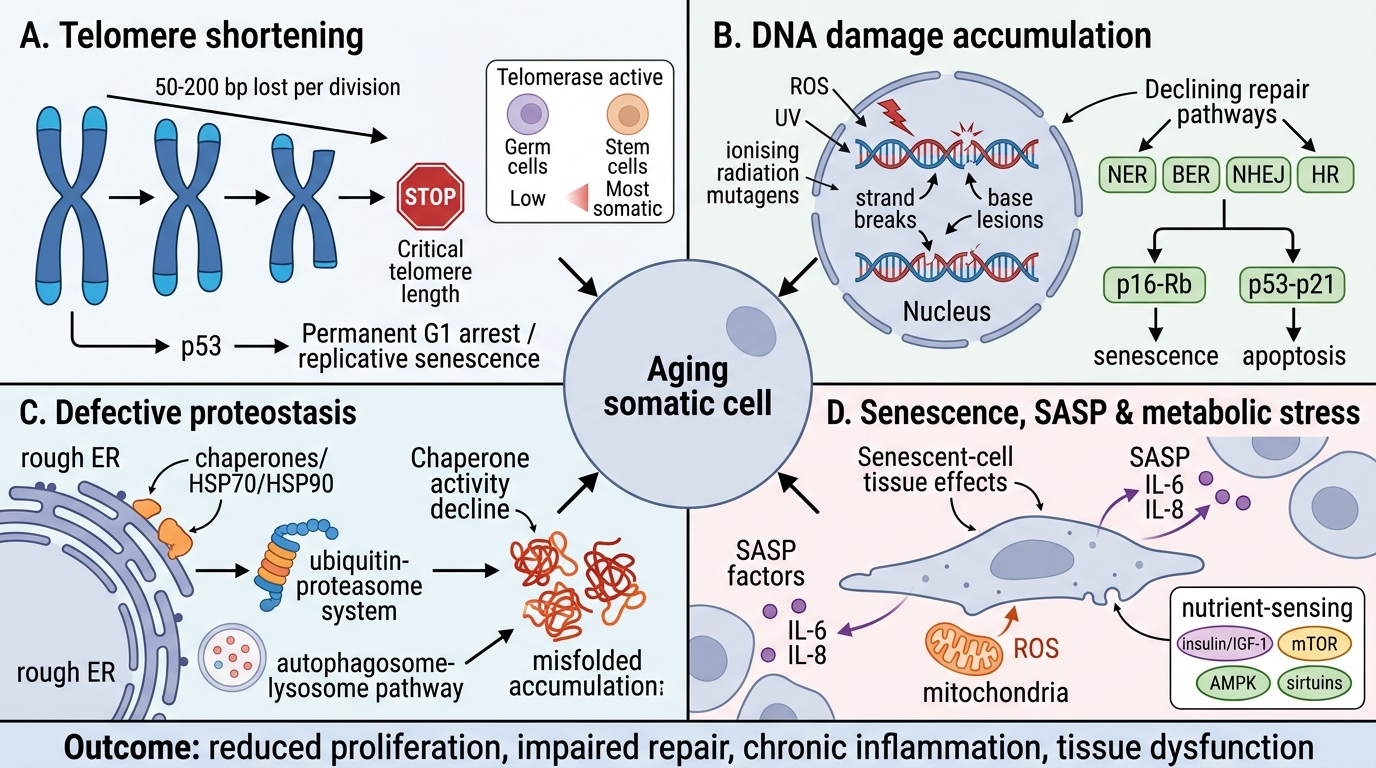
Cellular ageing refers to the progressive decline in cellular function and replicative capacity that occurs with chronological time and cumulative stress. It is driven by four overlapping mechanisms:
1. Telomere shortening:
Telomeres (TTAGGG repeats) protect chromosome ends. Each mitotic cycle shortens telomeres by 50–200 bp (end-replication problem). When telomeres reach a critical length, p53-mediated replicative senescence is triggered — cells enter permanent G1 arrest and cease dividing. Germ cells and stem cells maintain telomere length via telomerase (reverse transcriptase); most somatic cells do not.
Implication: Cells in tissues requiring high turnover (bone marrow, gut epithelium) accumulate telomere attrition faster. Premature ageing syndromes (Werner syndrome) involve defective DNA repair + accelerated telomere loss.
2. Accumulation of DNA damage:
ROS from mitochondrial respiration, UV, ionising radiation, and chemical mutagens cause cumulative DNA lesions (strand breaks, base modifications). Repair systems (NER, BER, NHEJ, HR) become progressively less efficient with age. Unrepaired damage activates p16-Rb and p53-p21 pathways → cellular senescence or apoptosis. Senescent cells secrete a senescence-associated secretory phenotype (SASP) — pro-inflammatory cytokines (IL-6, IL-8) — that propagate tissue dysfunction.
3. Defective protein homeostasis (proteostasis):
Misfolded proteins accumulate because:
• Chaperone activity (HSP70/HSP90) declines with age
• Proteasomal degradation efficiency falls
• Autophagy (the primary clearance mechanism for damaged organelles + protein aggregates) becomes impaired
Consequence: toxic protein aggregates accumulate — central to Alzheimer's disease (Aβ amyloid, tau), Parkinson's disease (α-synuclein Lewy bodies), Huntington's disease.
4. Decreased replicative capacity:
Stem cells undergo functional exhaustion — telomere attrition + epigenetic silencing → reduced self-renewal. The balance between pro-survival signals (mTOR, IGF-1) and pro-longevity pathways (SIRT1/FOXO3, caloric restriction → AMPK) shifts over time. Caloric restriction extends lifespan in model organisms by activating SIRT1 and AMPK, reducing mTOR signalling and enhancing autophagy.
Morphological correlates of ageing: nuclear pleomorphism, ↑lipofuscin (autophagy residue), irregular plasma membranes, loss of mitochondrial cristae, reduced rough ER.