Page 2 of 24
PA1.1-3,PA2.1-2 | Introduction to Pathology & Mechanisms of Cell Injury — SDL Guide (Part 2)
Cell Cycle, Proliferative Capacity, and Regenerative Medicine
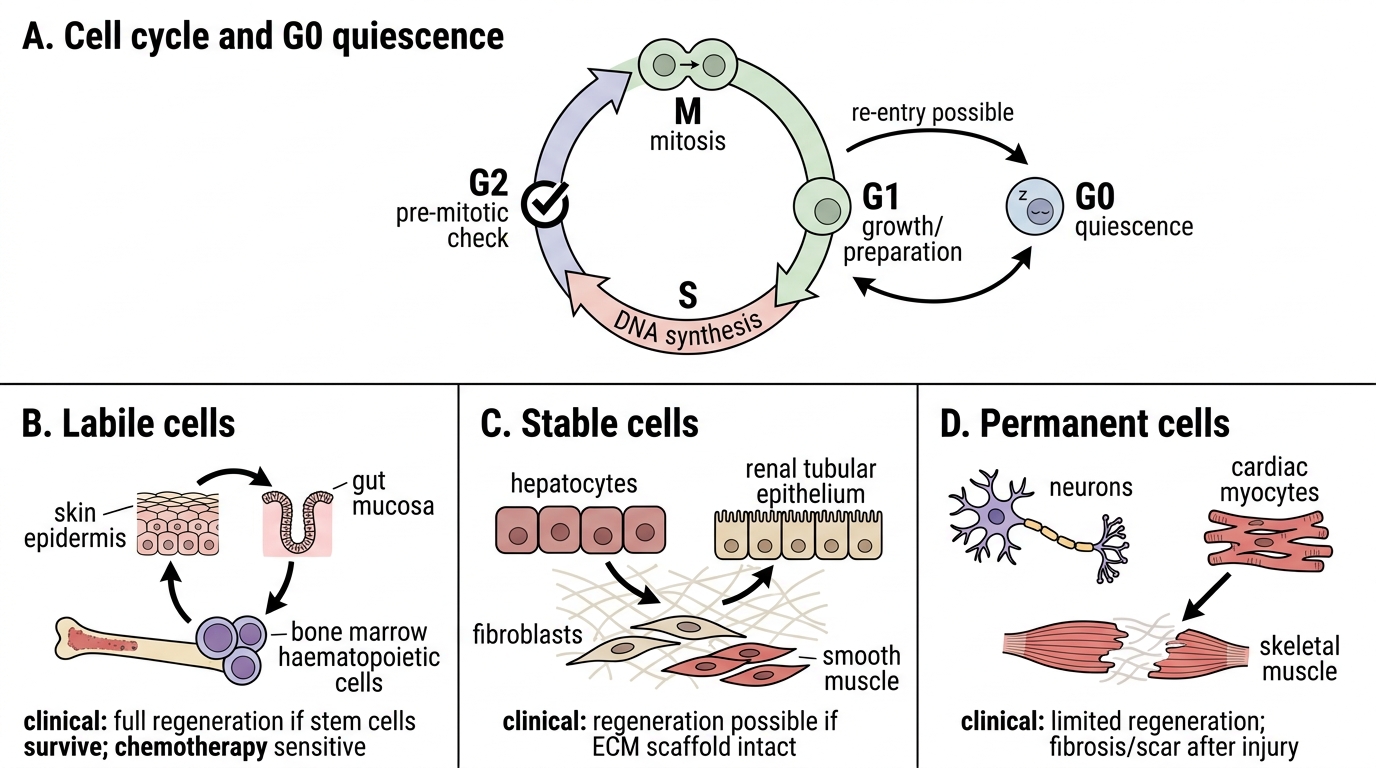
Cell Cycle and Tissue Regenerative Capacity
Recall that the cell cycle has four phases: G1 (growth/preparation), S (DNA synthesis), G2 (pre-mitotic check), and M (mitosis). Cells in G0 are quiescent but may re-enter the cycle.
The ability to regenerate after injury depends on proliferative capacity — tissues are classified as:
| Category | Definition | Examples | Pathological relevance |
|---|---|---|---|
| Labile cells | Continuously cycling; never leave the cycle | Skin epidermis, gut mucosa, bone marrow haematopoietic cells | Can regenerate fully if stem cells survive; also susceptible to chemotherapy |
| Stable cells | Normally quiescent (G0); re-enter cycle on demand | Hepatocytes, renal tubular epithelium, fibroblasts, smooth muscle | Can regenerate if extracellular matrix scaffold intact |
| Permanent cells | Terminally differentiated; cannot divide post-maturity | Cardiac myocytes, neurons, skeletal muscle | Irreversible loss; replaced by fibrosis (scar) |
Regenerative medicine leverages stem cells (embryonic, adult, induced pluripotent — iPSC) to replace permanently lost cells. Clinical applications include bone marrow transplantation (labile lineage), hepatocyte transplantation trials (stable lineage), and experimental cardiac patch technology (permanent lineage). The NMC competency PA1.3 expects awareness of this evolving field.
Causes of Cell Injury — The Etiological Spectrum
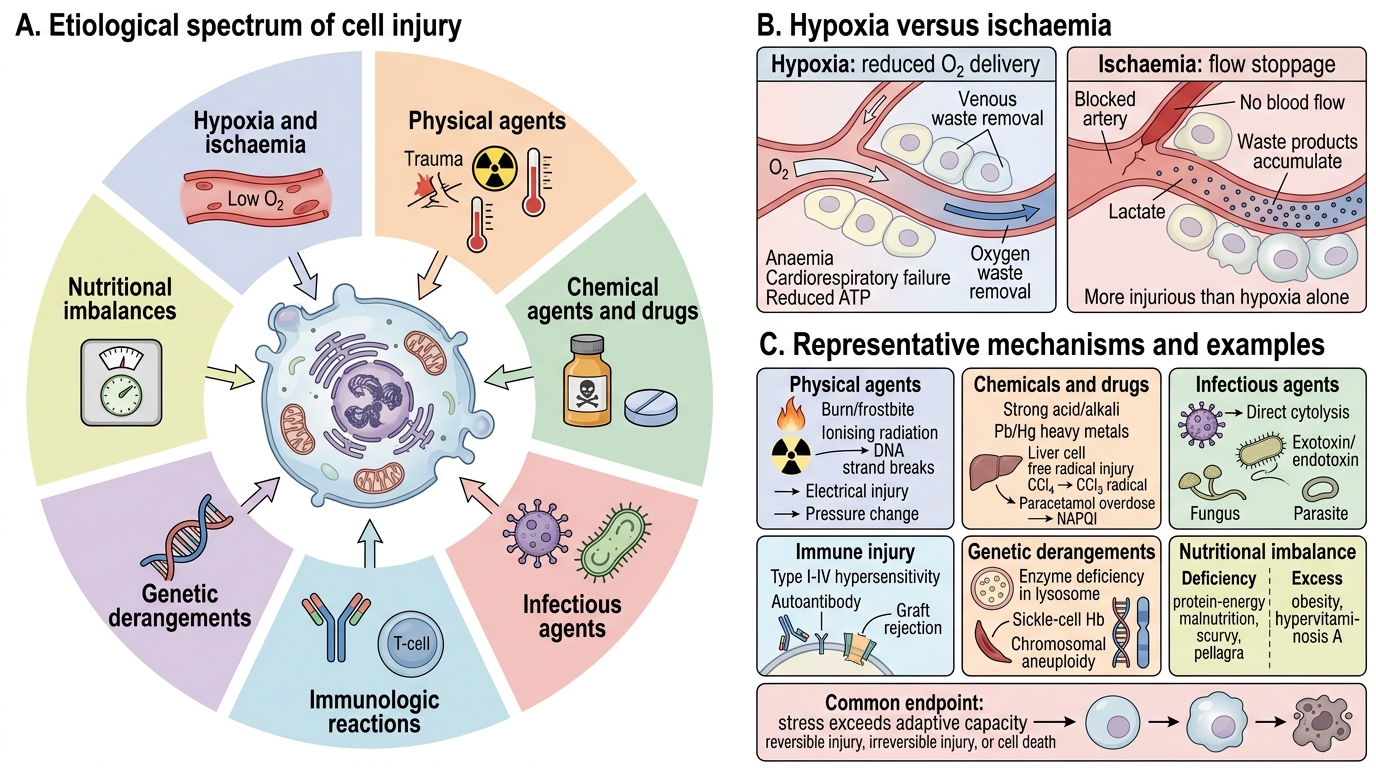
Causes of Cell Injury: Etiological Spectrum
Cell injury occurs when a stress exceeds the cell's adaptive capacity. The major categories are:
- Hypoxia and ischaemia — the most common cause; reduced O2 delivery (anaemia, cardiorespiratory failure) vs. complete flow stoppage (ischaemia). Ischaemia is more injurious than hypoxia alone because metabolic waste products also accumulate.
- Physical agents — mechanical trauma, extremes of temperature (burns, frostbite), ionising radiation (DNA strand breaks), sudden changes in atmospheric pressure, electrical injury.
- Chemical agents and drugs — direct toxicity (strong acids/alkalis, heavy metals: Pb, Hg) or indirect via reactive metabolites (carbon tetrachloride CCl₄ → •CCl₃ free radical; paracetamol overdose → NAPQI).
- Infectious agents — viruses (direct cytolysis or immune-mediated), bacteria (exotoxins, endotoxins), fungi, parasites.
- Immunologic reactions — hypersensitivity reactions (Type I–IV), autoimmune disease, graft rejection; immune-mediated injury can be as severe as direct infection.
- Genetic derangements — enzyme deficiencies (lysosomal storage disorders), structural protein defects (sickle-cell Hb), chromosomal aneuploidy.
- Nutritional imbalances — both deficiency (protein-energy malnutrition, scurvy, pellagra) and excess (obesity, hypervitaminosis A) cause cell injury.
Biochemical Mechanisms of Cell Injury — The Common Final Pathways
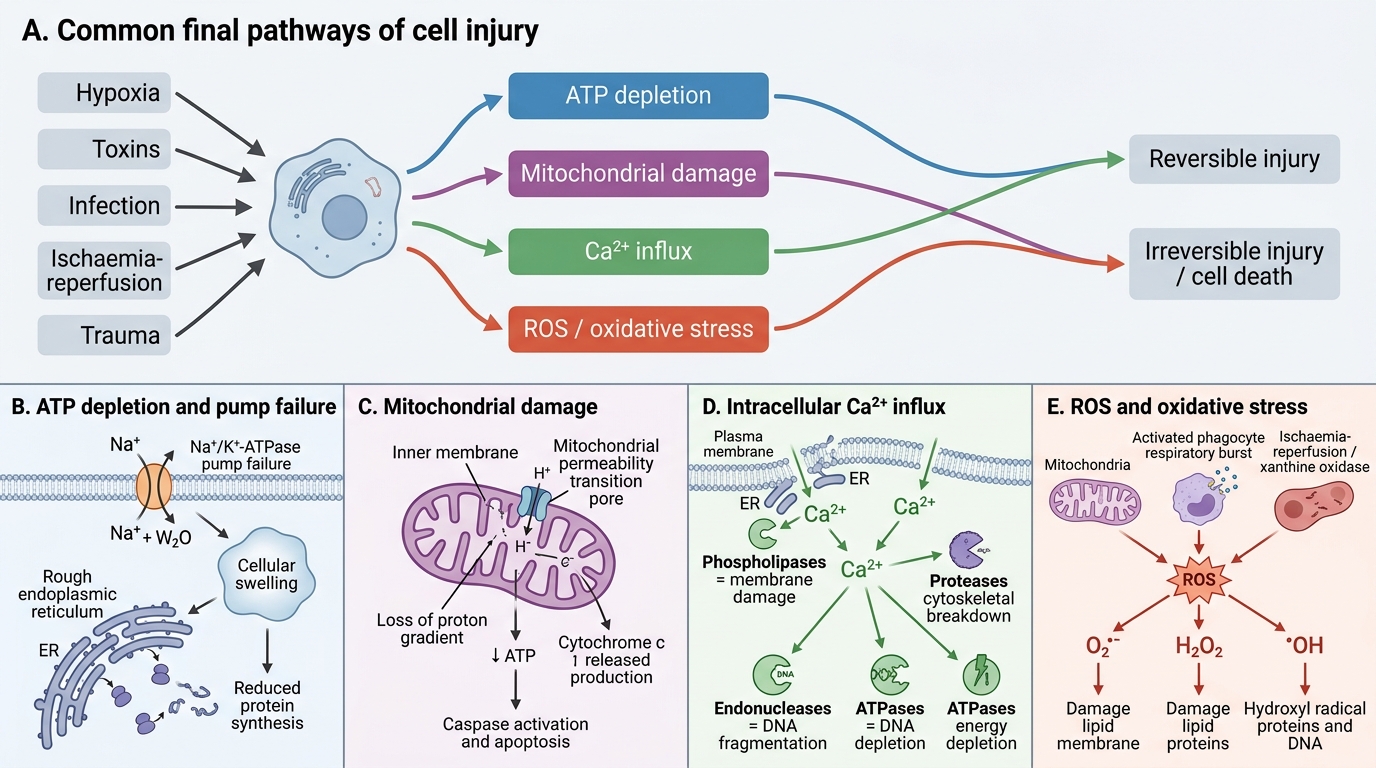
Common Final Pathways of Cell Injury
Regardless of the initial cause, cell injury tends to converge on a limited number of biochemical mechanisms. Understanding these is central to PA2.1.
1. ATP depletion
- ATP is consumed by the Na⁺/K⁺-ATPase pump, protein synthesis, lipogenesis, and membrane phospholipid turnover.
- Hypoxia → ↓oxidative phosphorylation → ↑anaerobic glycolysis → lactic acidosis + ↓ATP.
- Consequence: Na⁺ and water enter the cell (pump failure) → cellular swelling; ribosomes detach from ER → ↓protein synthesis.
2. Mitochondrial damage
- Mitochondrial dysfunction causes: (a) ↓ATP, (b) release of cytochrome c → caspase activation → apoptosis, (c) mitochondrial permeability transition (MPT) — an inner-membrane mega-channel opens, dissipating the proton gradient.
- MPT is a critical trigger for irreversible injury.
3. Intracellular calcium influx (↑[Ca²⁺]i)
- Normally, cytosolic Ca²⁺ is kept extremely low (~0.1 μM) by ER sequestration and plasma-membrane pumps.
- Membrane damage → massive Ca²⁺ entry → activates destructive enzymes: phospholipases (membrane damage), proteases (cytoskeletal/protein breakdown), endonucleases (DNA fragmentation), ATPases (↓energy).
4. Reactive oxygen species (ROS) and oxidative stress
- ROS include superoxide (O₂•⁻), hydrogen peroxide (H₂O₂), and hydroxyl radical (•OH).
- Sources: mitochondrial electron-transport leak, activated phagocytes (respiratory burst), ischaemia-reperfusion (xanthine oxidase), ionising radiation, drug metabolism.
- ROS cause: lipid peroxidation of membranes, protein cross-linking/oxidation, DNA strand breaks.
- Cellular defences: superoxide dismutase (SOD), catalase, glutathione peroxidase, vitamins C + E.
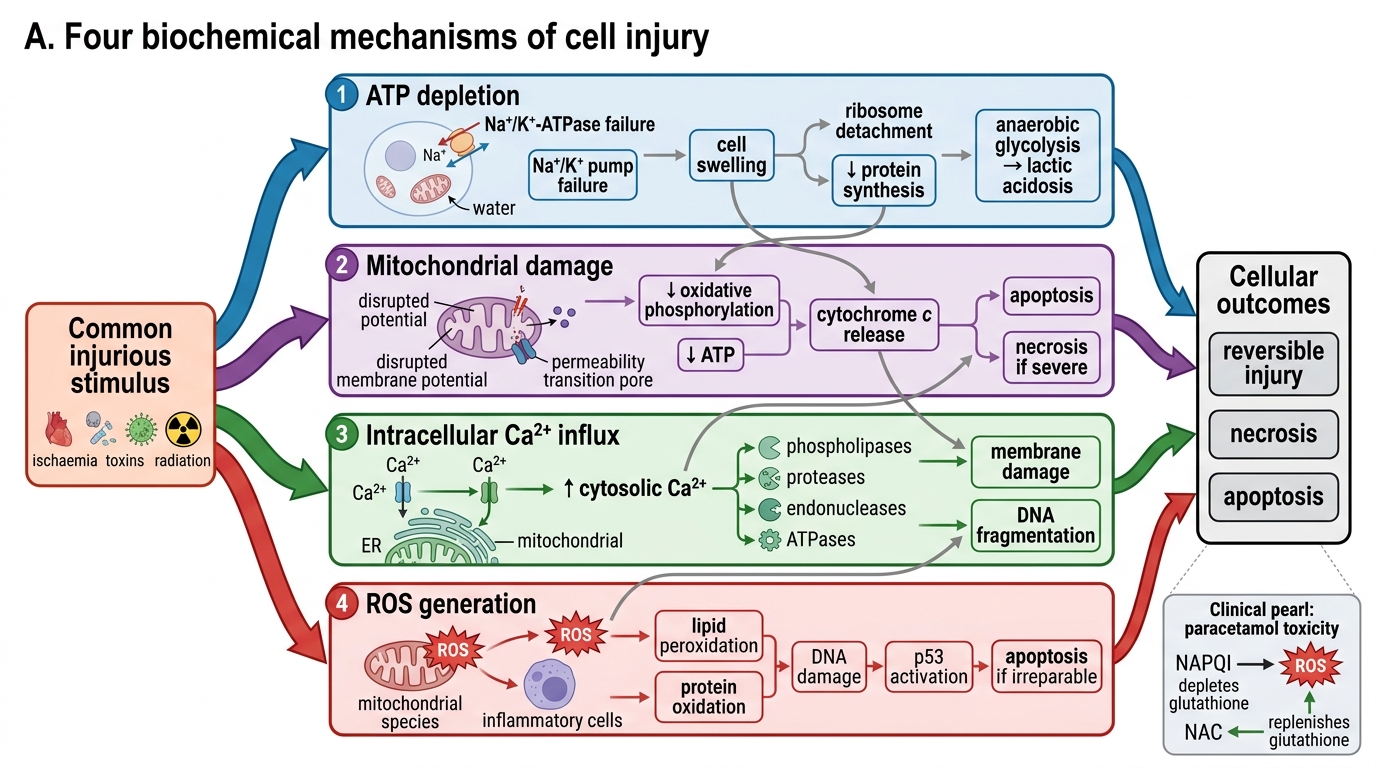
Four Biochemical Mechanisms of Cell Injury
5. Membrane damage
- Caused by: ↑[Ca²⁺]i-activated phospholipases, ROS lipid peroxidation, bacterial toxins (pore-forming), viral capsid proteins.
- The plasma membrane is the cell's boundary; lysosomal membrane rupture releases hydrolytic enzymes → autodigestion.
6. Protein misfolding and DNA damage
- Accumulation of misfolded proteins triggers endoplasmic reticulum stress → unfolded protein response (UPR) → if unresolved, pro-apoptotic signalling.
- DNA double-strand breaks (from ROS, radiation, replication errors) activate p53 → cell-cycle arrest → repair; if irreparable → apoptosis.
CLINICAL PEARL
N-acetylcysteine (NAC) in paracetamol overdose works by replenishing glutathione, the key intracellular antioxidant. Paracetamol's toxic metabolite NAPQI depletes glutathione → ROS accumulate → centrilobular hepatocyte necrosis. NAC given within 8–10 hours restores the ROS defence before irreversible injury occurs. This is biochemical mechanism translated directly to the bedside.
SELF-CHECK
Ischaemia-induced failure of the Na⁺/K⁺-ATPase pump leads to which immediate morphological change in the affected cell?
A. Nuclear pyknosis and chromatin condensation
B. Fatty change with lipid vacuole accumulation
C. Cellular swelling due to Na⁺ and water influx
D. Formation of myelin figures from membrane whorls
Reveal Answer
Answer: C. Cellular swelling due to Na⁺ and water influx
ATP depletion disables the Na⁺/K⁺-ATPase; intracellular Na⁺ rises, drawing in water osmotically. This produces cellular swelling (hydropic change) — the first and most common reversible morphological change. Nuclear pyknosis (A) is a feature of irreversible/necrotic injury. Fatty change (B) results from impaired lipoprotein secretion, not pump failure per se. Myelin figures (D) form later from membrane phospholipid breakdown.