Page 2 of 25
PA3.1-2 | Acute Inflammation — Vascular & Cellular Events, Mediators — SDL Guide (Part 2)
Leucocyte Recruitment Cascade
Leucocyte emigration from blood to tissue is a multistep adhesion cascade. It is predominantly neutrophils in the first 6–24 hours; monocytes predominate after 24–48 hours.
Step 1 — Margination: As stasis develops, neutrophils move from the axial stream to the vessel periphery.
Step 2 — Rolling: Selectins mediate loose, reversible tethering:
• P-selectin: stored in Weibel–Palade bodies of endothelium; rapidly mobilised by histamine.
• E-selectin: induced on endothelium by IL-1 and TNF (hours later).
• L-selectin: constitutively expressed on leucocytes.
Selectins bind glycoprotein ligands (e.g., P-selectin glycoprotein ligand-1, PSGL-1).
Step 3 — Activation: Chemokines (e.g., IL-8/CXCL8) displayed on endothelial surface bind G-protein-coupled receptors on rolling neutrophils → signal transduction → integrin activation.
Step 4 — Firm adhesion: Activated integrins (LFA-1 / Mac-1; CD11a–CD18 / CD11b–CD18) bind ICAM-1 (intercellular adhesion molecule-1) and VCAM-1 on endothelium. This is high-affinity, stable arrest.
Step 5 — Transmigration (diapedesis): Neutrophils squeeze between endothelial cells (paracellular) at the junction, guided by PECAM-1 (CD31). They then cross the basement membrane using matrix metalloproteinases (collagenase).
Step 6 — Chemotaxis: Directional migration toward the injury site along a chemical gradient. Major chemotactic agents:
• C5a (complement)
• LTB₄ (leukotriene B4)
• IL-8 / CXCL8 (chemokine)
• Bacterial products (e.g., N-formyl-methionyl peptides, f-Met-Leu-Phe)
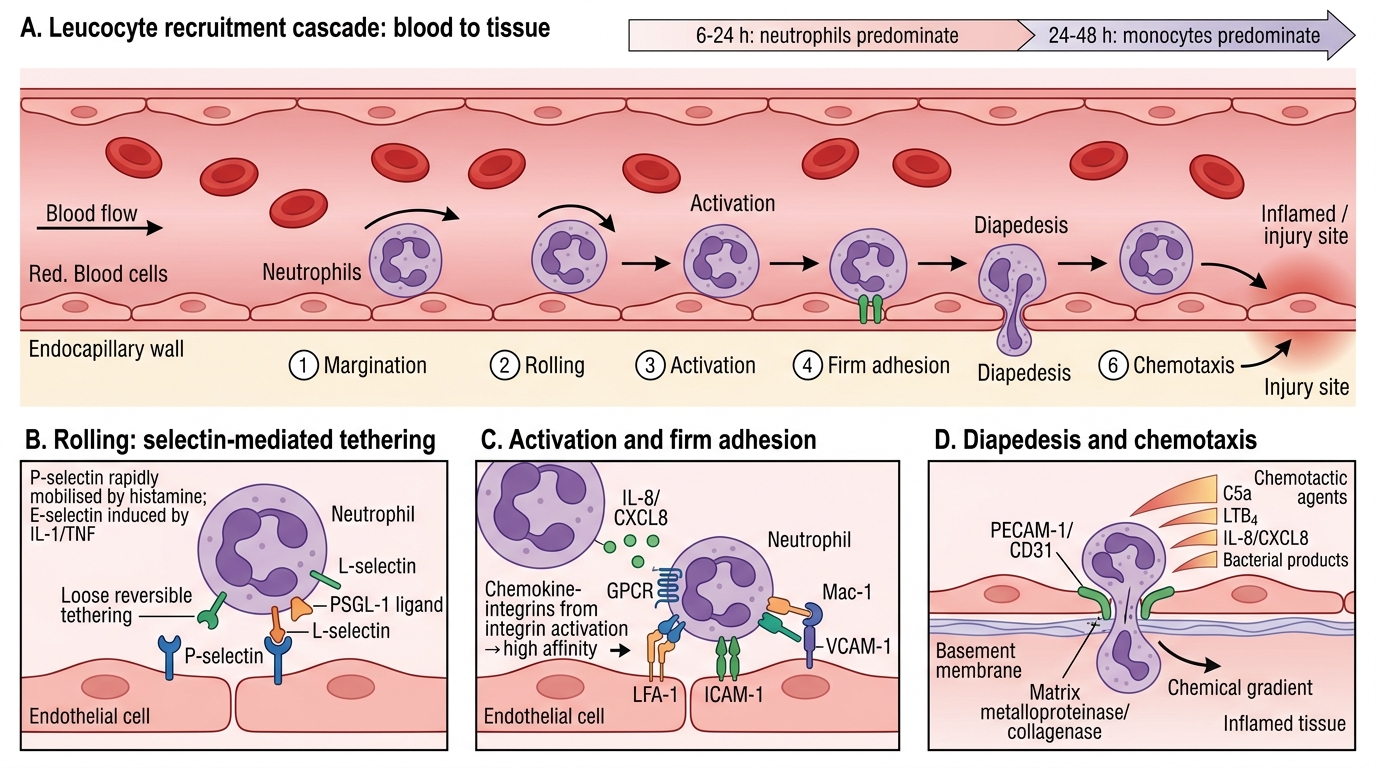
Leucocyte Recruitment Cascade
CLINICAL PEARL
Leucocyte adhesion deficiency (LAD) is a rare autosomal recessive disorder caused by mutations in the CD18 gene, resulting in absent or dysfunctional β₂-integrins (LFA-1, Mac-1). Affected children suffer recurrent bacterial infections with markedly elevated blood neutrophil counts (neutrophilia) but NO pus formation at infection sites — because neutrophils cannot undergo firm adhesion or diapedesis. This directly illustrates why integrin–ICAM-1 binding is essential for effective leucocyte emigration.
Phagocytosis and Microbicidal Mechanisms
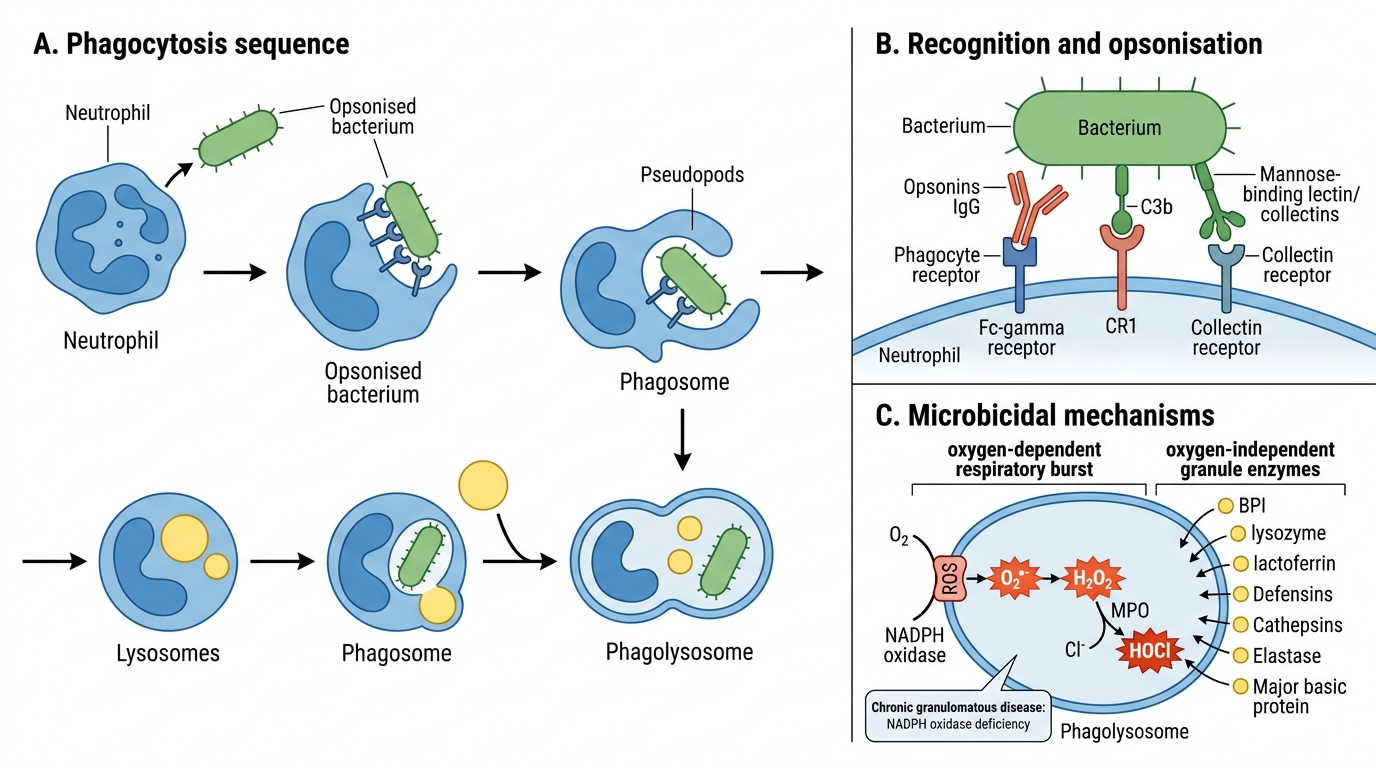
Phagocytosis and Microbicidal Killing
Once at the site, neutrophils (and macrophages) destroy pathogens via phagocytosis.
Phase 1 — Recognition and opsonisation:
Opsonins coat pathogens and enhance phagocyte binding:
• IgG Fc fragment — binds Fc-γR on phagocytes.
• C3b (complement) — binds CR1 (complement receptor 1).
• Collectins (mannose-binding lectin).
Phase 2 — Engulfment: Pseudopods extend and fuse around the particle → phagosome. The phagosome fuses with lysosomes → phagolysosome.
Phase 3 — Killing:
Oxygen-dependent (respiratory burst / oxidative burst):
• NADPH oxidase complex on the phagosome membrane: O₂ → superoxide (O₂•⁻) via electron transfer.
• Superoxide → H₂O₂ (hydrogen peroxide) spontaneously or via SOD.
• Myeloperoxidase (MPO) catalyses: H₂O₂ + Cl⁻ → hypochlorous acid (HOCl) — the most potent bactericidal agent in neutrophils.
• Also: •OH (hydroxyl radical), singlet O₂, NO.
Oxygen-independent:
• Bactericidal/permeability-increasing protein (BPI): disrupts gram-negative outer membrane.
• Lysozyme: cleaves peptidoglycan of gram-positive bacteria.
• Lactoferrin: chelates iron (bacteria need iron).
• Major basic protein: kills parasites (eosinophils).
• Defensins, cathepsins, elastase (from azurophil granules).
> Chronic granulomatous disease (CGD) — NADPH oxidase deficiency → defective respiratory burst → recurrent catalase-positive organism infections (Staph, Aspergillus).
SELF-CHECK
A child presents with recurrent deep-seated bacterial infections. His blood count shows marked neutrophilia, but wound biopsies show NO neutrophil infiltrate. Which molecule is most likely defective?
A. P-selectin on endothelial cells
B. CD18 (β₂-integrin subunit)
C. Myeloperoxidase in neutrophil granules
D. C3b complement fragment
Reveal Answer
Answer: B. CD18 (β₂-integrin subunit)
The clinical picture — high circulating neutrophils with absent tissue infiltration — is classic for Leucocyte Adhesion Deficiency (LAD), caused by CD18 (β₂-integrin) deficiency. Without functional LFA-1/Mac-1, neutrophils cannot achieve firm adhesion to ICAM-1 on endothelium and cannot transmigrate. P-selectin deficiency would impair rolling (less dramatic). MPO deficiency impairs killing, not migration. C3b deficiency affects opsonisation and chemotaxis.
Chemical Mediators — Overview
Mediators are the molecular language of acute inflammation. They are derived from plasma (inactive precursors → activated) or produced by cells (pre-formed or newly synthesised). Key principle: most mediators have short half-lives, are quickly inactivated, or have built-in counter-regulatory mechanisms — preventing excessive injury.
Cell-derived mediators:
• Vasoactive amines (pre-formed, immediate release)
• Arachidonic acid metabolites (newly synthesised, minutes)
• Platelet-activating factor (cell membrane–derived)
• Cytokines (synthesised de novo, hours)
• Reactive oxygen species and nitric oxide
Plasma-derived mediators (activated in plasma):
• Complement fragments
• Kinin system
• Coagulation/fibrinolytic system
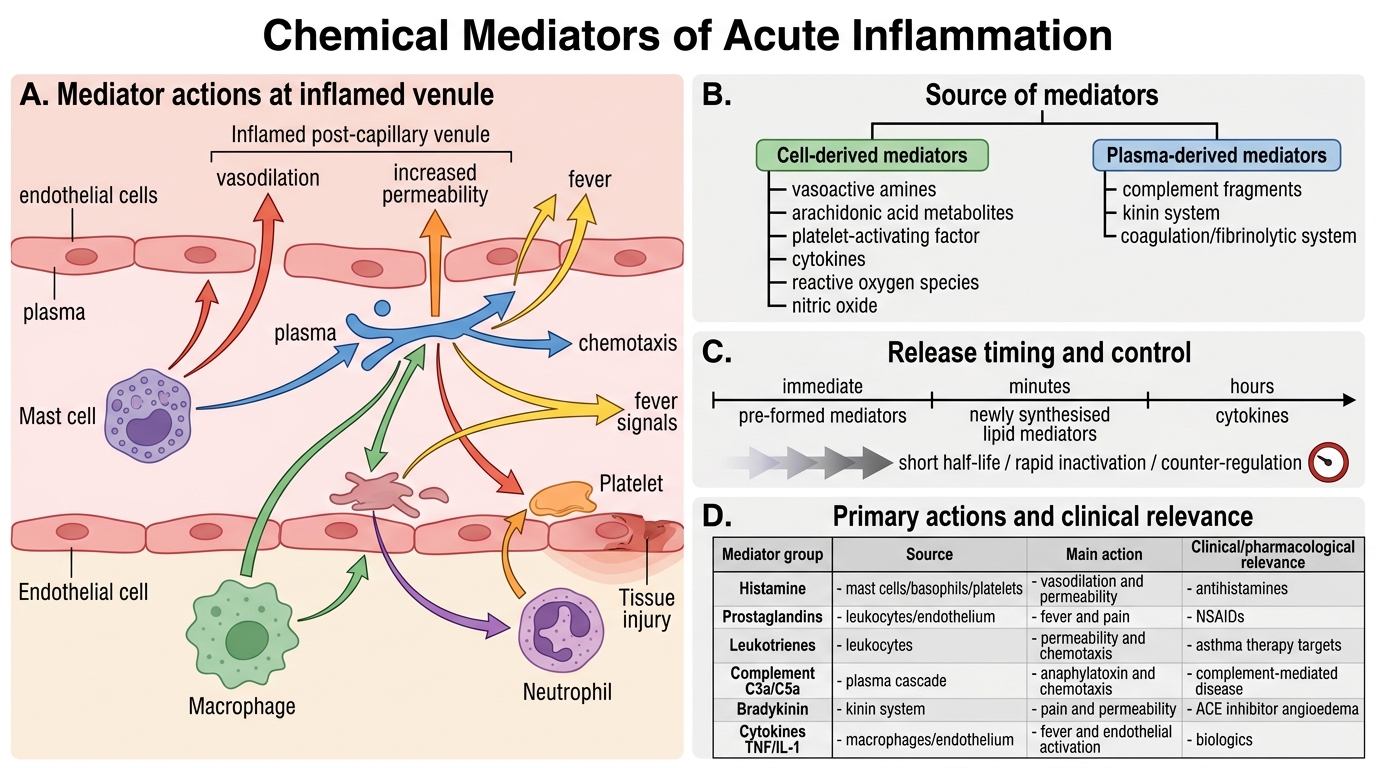
Chemical Mediators of Acute Inflammation