Page 11 of 24
PA6.4-6 | Tumour Effects, Immunology & Laboratory Diagnosis — SDL Guide (Part 2)
Tumour Immunology — Antigens and Surveillance
⚑ AI image — pending faculty review (auto-QA score 6/10; best of 3 attempts)
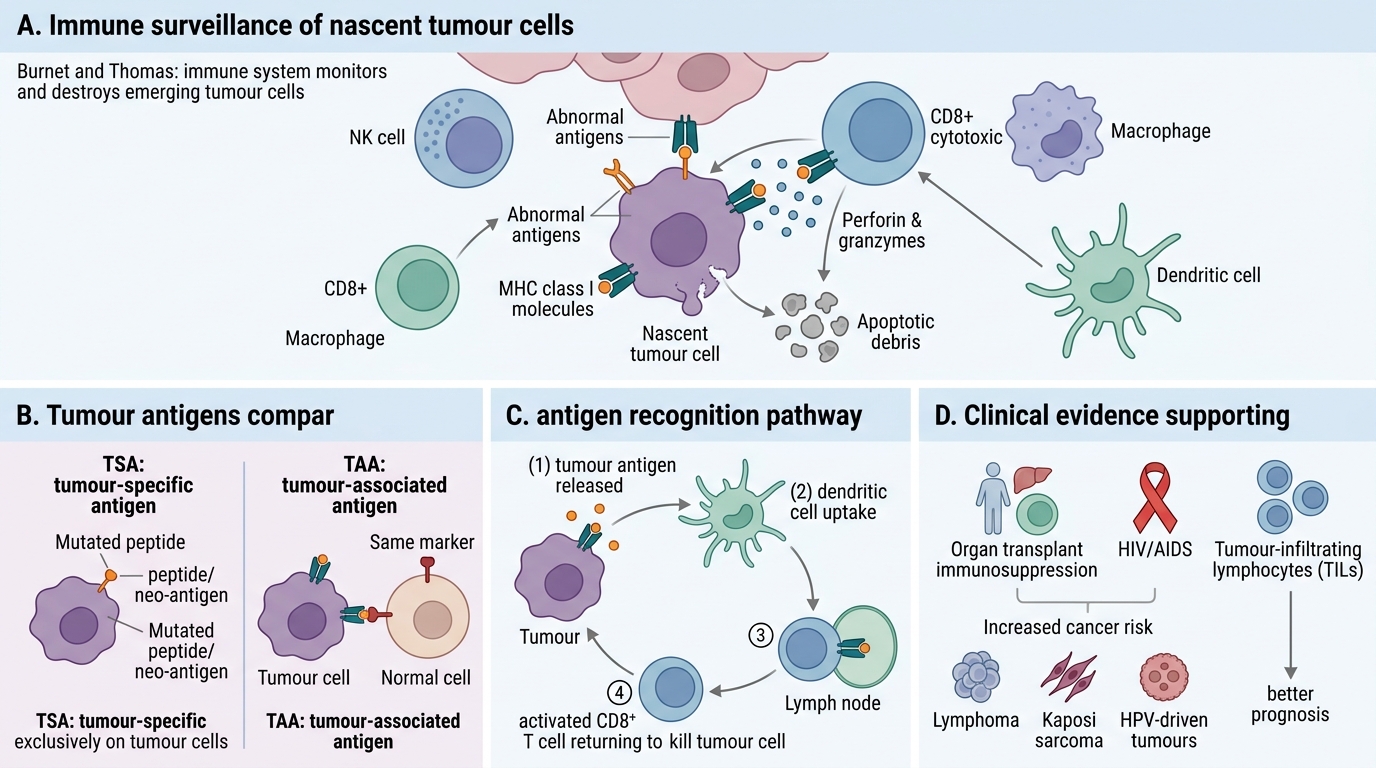
Tumour Immunology: Antigens and Immune Surveillance
The immune system continuously monitors for and destroys nascent tumour cells — a process called immune surveillance (proposed by Burnet and Thomas). Clinical evidence:
- Immunosuppressed patients (organ transplants, HIV/AIDS) have dramatically higher rates of certain cancers (lymphomas, Kaposi sarcoma, HPV-driven tumours).
- Tumour-infiltrating lymphocytes (TILs) correlate with better prognosis in many cancers.
Tumour antigens — molecules recognised as non-self by immune cells — fall into two broad classes:
| Class | Full name | Basis | Examples |
|---|---|---|---|
| TSA | Tumour-specific antigen | Present ONLY on tumour cells | Neo-antigens from somatic mutations (unique to each tumour) |
| TAA | Tumour-associated antigen | Over-expressed or aberrantly expressed on tumour; also found on normal cells | AFP (normally fetal), PSA, CEA, HER2 |
Effector mechanisms of anti-tumour immunity:
- Cytotoxic T lymphocytes (CTLs): CD8+ T cells recognise tumour peptides on MHC class I → perforin/granzyme-mediated killing. The primary adaptive mechanism.
- NK cells: Kill tumour cells that have down-regulated MHC I (a common evasion strategy — NK cells are activated by 'missing self').
- Antibody-dependent cellular cytotoxicity (ADCC): Tumour-specific antibodies opsonise tumour cells → Fc-receptor-mediated killing by NK cells and macrophages.
- Macrophages (M1): Activated macrophages release ROS and NO, directly cytotoxic.
Immune Evasion by Tumours
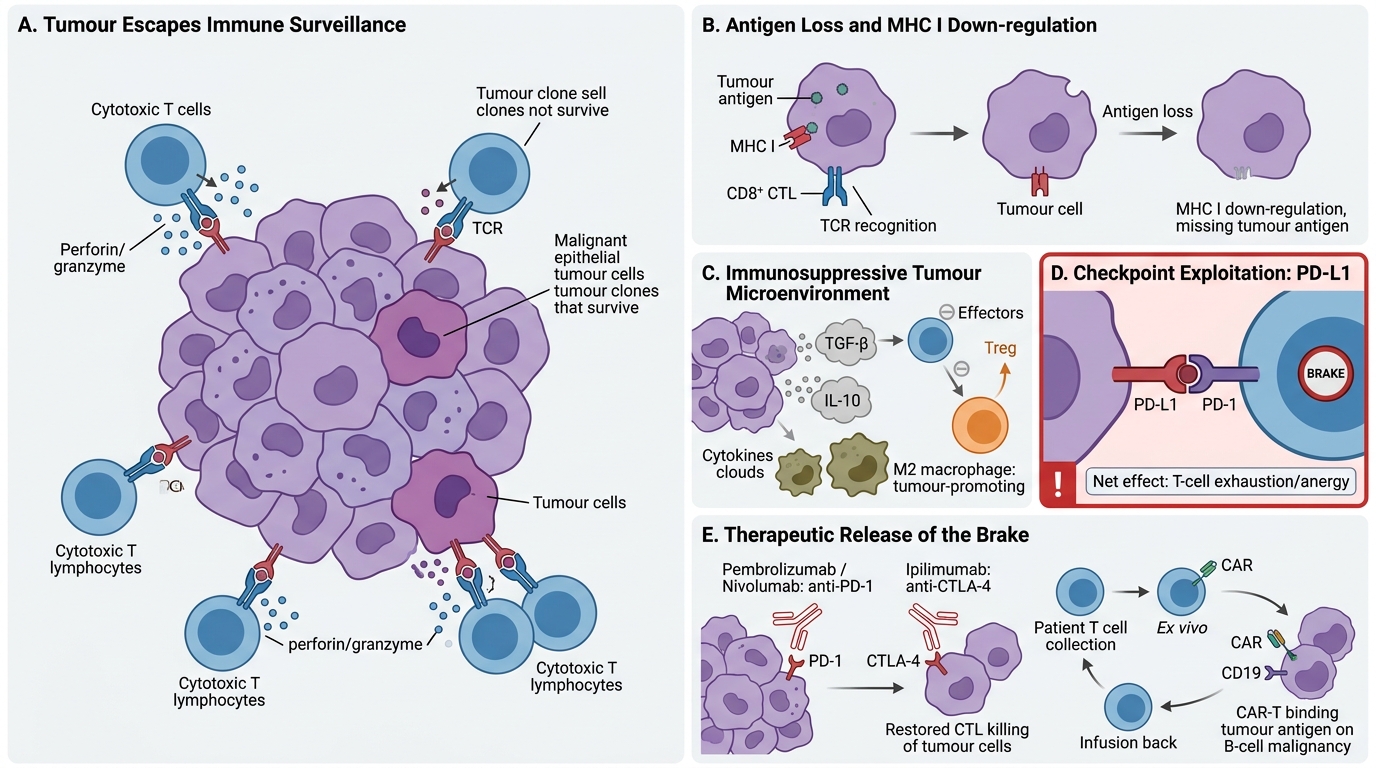
Immune Evasion by Tumours
Tumours that succeed clinically have escaped immune surveillance. Key evasion mechanisms:
- Antigen loss / down-regulation: Tumour cells with high immunogenicity are selectively killed (immunoediting); surviving clones have lost or down-regulated MHC I or tumour antigens — so CTLs cannot recognise them.
- Immunosuppressive microenvironment: Tumours secrete TGF-β and IL-10, which suppress T-cell activation and convert effector T cells into regulatory T cells (Tregs). The tumour micro-environment (TME) is rich in Tregs and M2 macrophages (tumour-promoting).
- Checkpoint exploitation — PD-L1: Many tumours up-regulate PD-L1 (Programmed Death Ligand 1) on their surface. PD-L1 binds PD-1 on T cells → T-cell exhaustion/anergy. This is the most clinically important evasion mechanism because it is directly targetable.
Basis of immunotherapy:
- Checkpoint inhibitors: Monoclonal antibodies that block PD-1 (pembrolizumab, nivolumab) or CTLA-4 (ipilimumab) → release T-cell brake → restore anti-tumour killing. Used in melanoma, NSCLC, bladder cancer, and others.
- CAR-T cell therapy: Patient's T cells are genetically engineered ex vivo to express a chimeric antigen receptor (CAR) targeting a tumour antigen (e.g., CD19 in B-cell lymphoma/ALL). Highly effective in select haematologic malignancies.
SELF-CHECK
A tumour up-regulates PD-L1 on its surface. What is the net immunological effect on tumour-reactive T cells?
A. Enhanced CTL proliferation via co-stimulation
B. T-cell exhaustion and anergy via PD-1 signalling
C. Increased NK cell recruitment and ADCC
D. Upregulation of MHC class I presentation
Reveal Answer
Answer: B. T-cell exhaustion and anergy via PD-1 signalling
PD-L1 on the tumour surface binds PD-1 on T cells, delivering an inhibitory signal that causes T-cell exhaustion and functional anergy. This is the mechanistic basis for using PD-1/PD-L1 checkpoint inhibitors (pembrolizumab, nivolumab) to restore anti-tumour immunity.
Laboratory Diagnosis — Histopathology and Cytology
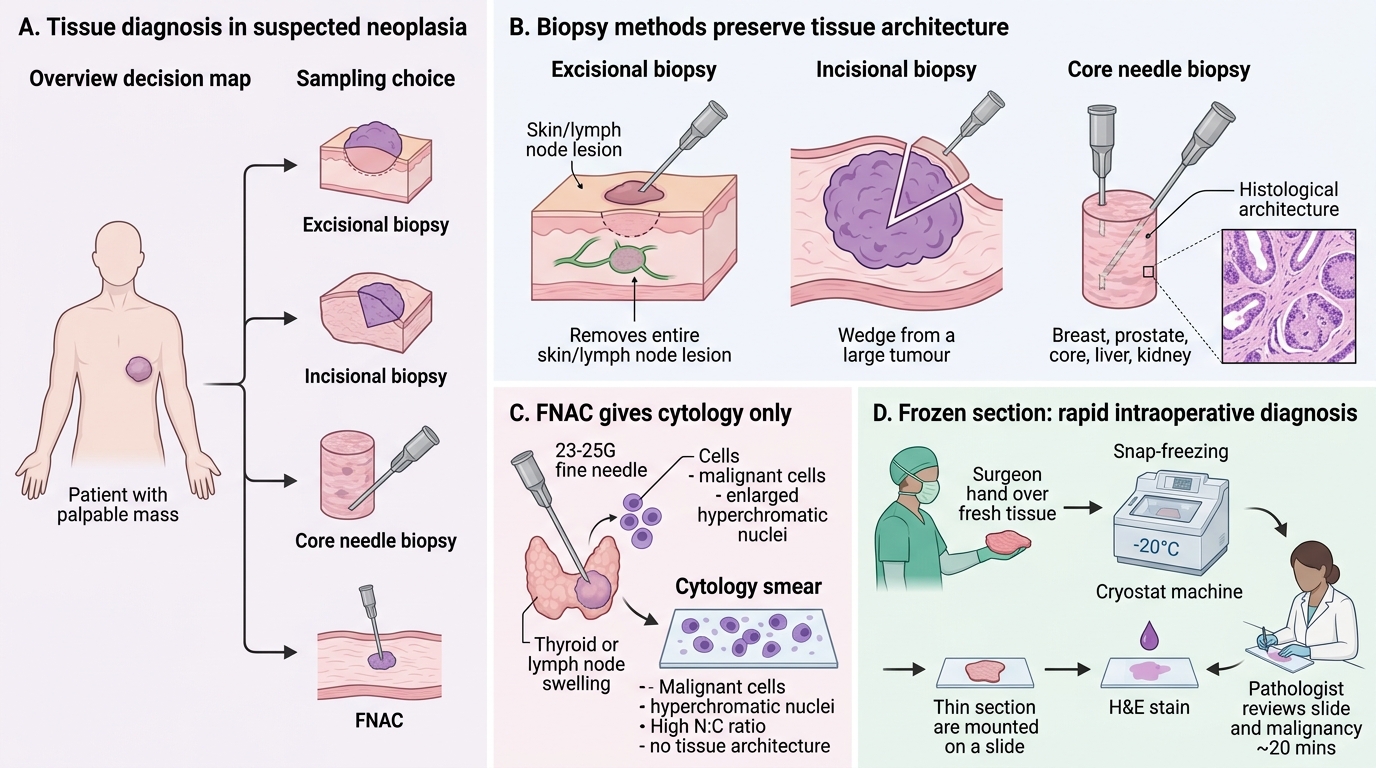
Tissue Sampling Methods in Cancer Diagnosis
Definitive cancer diagnosis requires tissue. The choice of sampling technique depends on lesion accessibility, required material, and urgency.
Biopsy types:
| Type | Description | Best for |
|---|---|---|
| Excisional biopsy | Entire lesion removed | Small, accessible lesions (skin, lymph node) |
| Incisional biopsy | Wedge from large lesion | Large soft-tissue tumours; when excision not feasible |
| Core needle biopsy (Tru-Cut) | Hollow-needle core of tissue | Breast, prostate, liver, kidney — preserves architecture |
| FNAC (Fine Needle Aspiration Cytology) | 23–25G needle, cytological smear | Rapid diagnosis (thyroid, breast, lymph node, salivary gland); cytology only, no architecture |
Frozen section: Intraoperative technique — tissue snap-frozen, cut at −20°C, H&E stained in ~20 min. Used to:
1. Confirm malignancy intraoperatively (dictates extent of resection).
2. Assess surgical margins (clear vs involved).
3. Identify lymph node involvement.
Limitation: ice crystal artefact reduces cytological detail; not for all specimen types.
Cytology (non-biopsy):
• Exfoliative cytology: Cervical Pap smear, sputum, urine, CSF, pleural/peritoneal fluid.
• Imprint cytology: Touch preparation of cut biopsy surface.
Routine histopathology (H&E) identifies tissue architecture + cellular features (pleomorphism, mitoses, necrosis, invasion) — the gold standard for diagnosis and grading.