Page 6 of 22
PA8.3-5 | HLA, Transplantation, Autoimmunity & SLE — SDL Guide (Part 2)
Autoimmunity — Definition, Tolerance, and Its Failure
Autoimmunity: Failure of Self-Tolerance
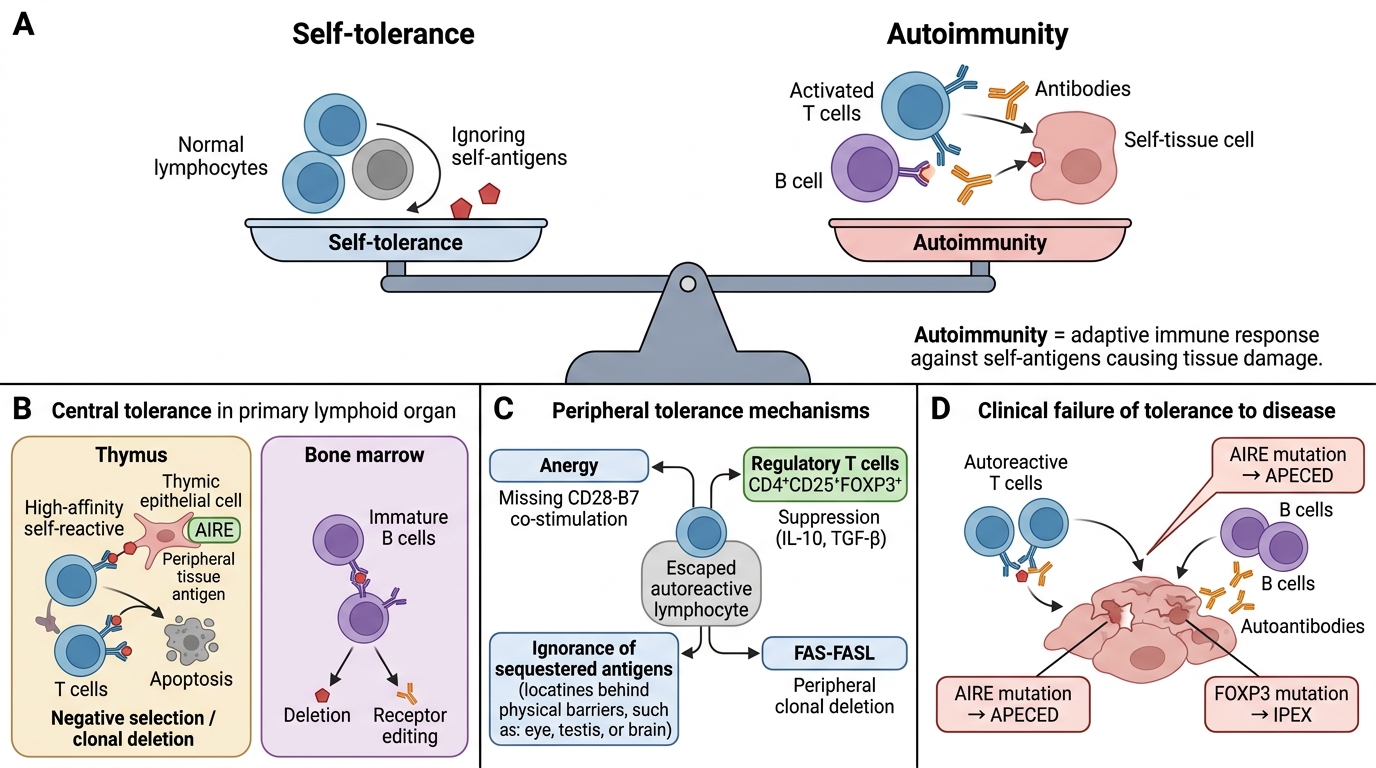
Autoimmunity is a state in which an adaptive immune response (T cells, B cells, or antibodies) is directed against self-antigens, causing tissue damage. It is the pathological counterpart of self-tolerance — the normal state in which the immune system is unresponsive to self.
Central tolerance (in primary lymphoid organs):
• T cells (thymus): During thymic development, T cells whose TCRs bind self-peptide–MHC with high affinity are deleted by apoptosis (clonal deletion / negative selection). The gene AIRE (autoimmune regulator) in thymic epithelium drives expression of peripheral tissue antigens so potentially autoreactive T cells can be eliminated. AIRE mutations → APECED syndrome (autoimmune polyendocrinopathy).
• B cells (bone marrow): Immature B cells binding self-antigen are either deleted or undergo receptor editing (rearrange BCR to escape autoreactivity).
Peripheral tolerance mechanisms (for cells that escape central deletion):
1. Anergy — autoreactive T cells in periphery receive TCR signal without co-stimulation (CD28–B7) → become unresponsive.
2. Regulatory T cells (T_reg, CD4+CD25+FOXP3+) — suppress autoreactive T and B cells via IL-10, TGF-β, and direct contact. FOXP3 mutations → IPEX syndrome (immune dysregulation, polyendocrinopathy).
3. Ignorance — self-antigens sequestered behind barriers (eye, testis, brain) are never encountered by lymphocytes.
4. Peripheral clonal deletion — FAS–FASL-mediated apoptosis of chronically stimulated autoreactive cells.
Mechanisms of tolerance failure (pathways to autoimmunity):
1. Molecular mimicry — microbial antigen shares structural similarity with self-antigen. Immune response to microbe cross-reacts with self. Classic example: Group A Streptococcus M protein epitopes → anti-myosin antibodies → rheumatic heart disease.
2. Bystander activation — inflammation at a site releases sequestered self-antigens or activates APCs → autoreactive T cells receive sufficient co-stimulation to become activated.
3. Epitope spreading — initial immune response to a dominant self-epitope; tissue damage releases other self-antigens → new autoreactive responses broaden the attack.
4. Loss of T_reg function — insufficient suppression allows autoreactive clones to expand.
5. Genetic factors — certain HLA alleles (DR3, DR4, B27), polymorphisms in CTLA4 (co-stimulation checkpoint), PTPN22 (T-cell signalling), and complement genes (C1q deficiency → impaired immune complex clearance → SLE susceptibility).
6. Environmental triggers — UV light (releases nuclear antigens in keratinocytes — relevant to SLE flares), viral infections (EBV activates polyclonal B cells), drugs (procainamide, hydralazine → drug-induced lupus).
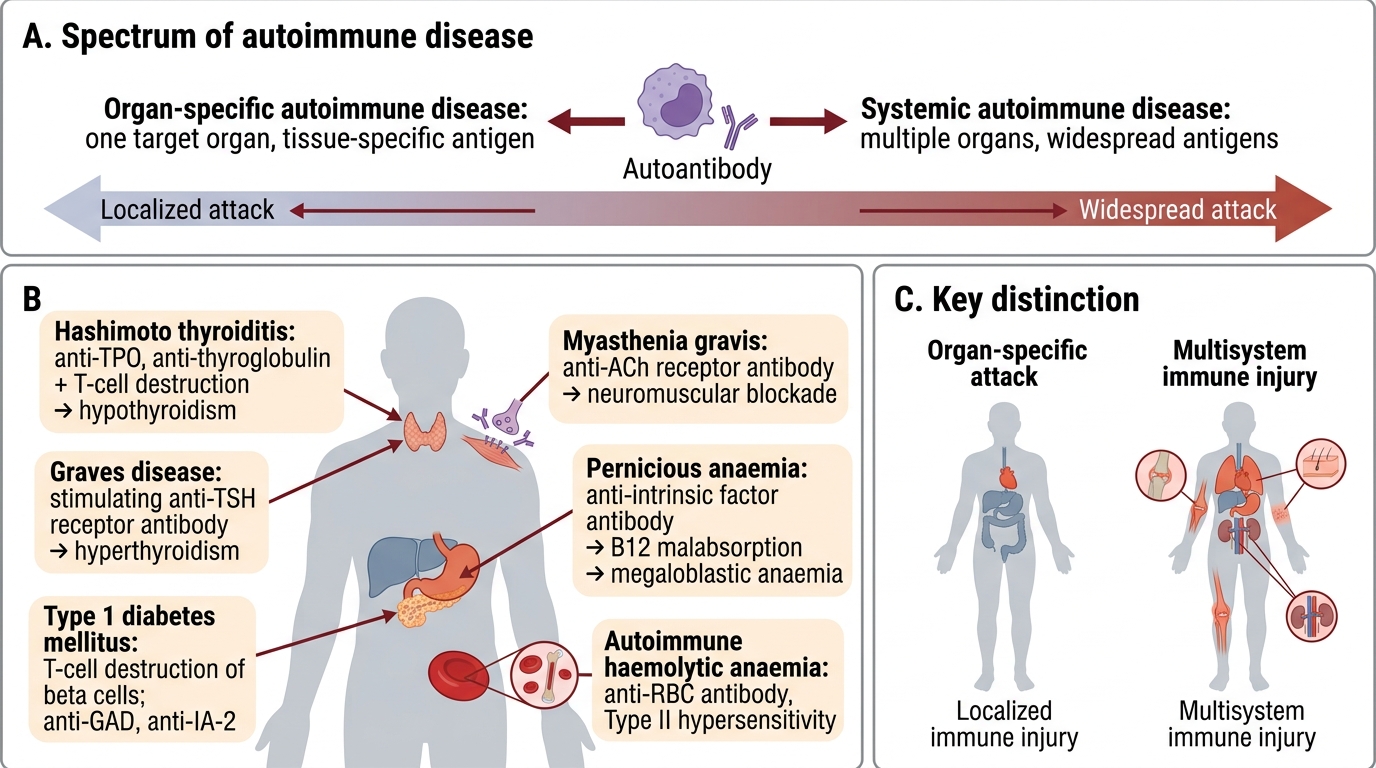
Spectrum of Autoimmune Diseases — Organ-Specific vs Systemic
Autoimmune diseases are broadly classified by the distribution of the immune attack:
Organ-specific autoimmune diseases — damage confined to one target organ; autoantibody often directed at a tissue-specific antigen:
| Disease | Target antigen / mechanism |
|---|---|
| Hashimoto thyroiditis | Anti-TPO, anti-thyroglobulin antibodies + T-cell destruction → hypothyroidism |
| Graves disease | Anti-TSH-receptor stimulating antibody → hyperthyroidism |
| Type 1 diabetes mellitus | T-cell destruction of islet β-cells; anti-GAD, anti-IA-2 antibodies |
| Myasthenia gravis | Anti-acetylcholine receptor antibodies → neuromuscular blockade |
| Pernicious anaemia | Anti-intrinsic factor antibodies → vitamin B12 malabsorption → megaloblastic anaemia |
| Autoimmune haemolytic anaemia | Anti-RBC antibodies (Type II hypersensitivity) |
Systemic autoimmune diseases — multiple organs affected; autoantibodies often directed at ubiquitous antigens:
| Disease | Hallmark feature |
|---|---|
| SLE | Anti-dsDNA, anti-Sm; immune-complex deposition; multisystem |
| Rheumatoid arthritis | Anti-CCP, RF; synovitis → cartilage destruction |
| Systemic sclerosis (scleroderma) | Anti-Scl-70 (diffuse), anti-centromere (limited); fibrosis + vasculopathy |
| Sjögren syndrome | Anti-Ro (SSA), anti-La (SSB); exocrine gland destruction → sicca |
| Polymyositis / dermatomyositis | Anti-Jo-1; muscle inflammation |
| Mixed connective tissue disease | Anti-U1 RNP; overlap features |
A key point: organ-specific diseases tend to be T-cell + organ-directed antibody mediated; systemic diseases tend to involve immune-complex deposition and anti-nuclear antibodies.
Spectrum of Autoimmune Diseases
SELF-CHECK
A 35-year-old woman develops progressive proximal muscle weakness. Serology shows anti-Jo-1 antibodies. The most likely diagnosis is:
A. Systemic lupus erythematosus
B. Sjögren syndrome
C. Polymyositis
D. Myasthenia gravis
Reveal Answer
Answer: C. Polymyositis
Anti-Jo-1 antibodies are the classic serological marker of polymyositis (and dermatomyositis). Proximal muscle weakness is the hallmark clinical feature. SLE is associated with anti-dsDNA and anti-Sm; Sjögren with anti-Ro/La; myasthenia gravis with anti-acetylcholine receptor antibodies.
SLE — Pathogenesis
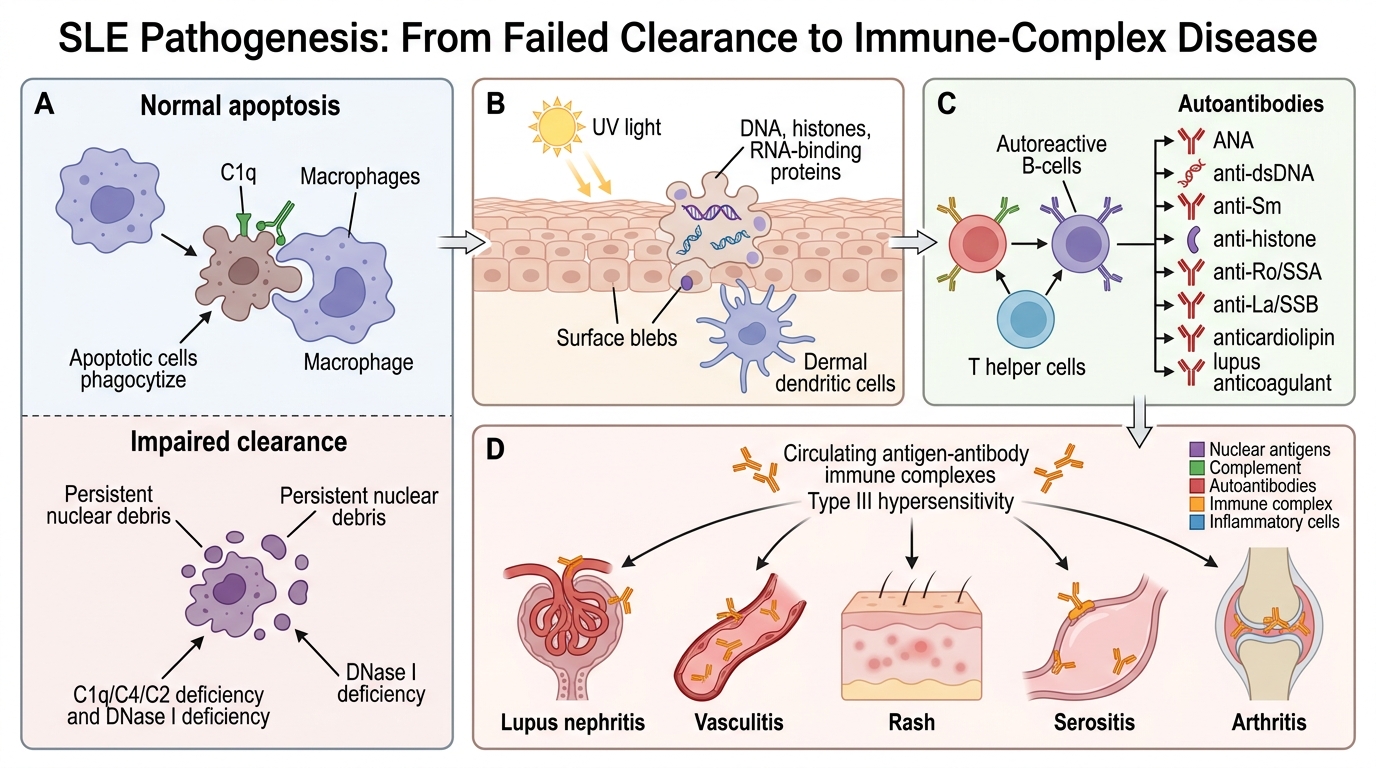
Systemic lupus erythematosus (SLE) is the prototypic systemic autoimmune disease, driven by a fundamental failure to clear apoptotic cell debris, leading to sustained autoantibody production and immune-complex deposition across multiple organ systems.
Step 1 — Loss of tolerance to nuclear antigens
Normal apoptosis generates nuclear fragments (DNA, histones, RNA-binding proteins) that are rapidly cleared by macrophages via C1q-mediated phagocytosis. In SLE:
• Genetic defects — complement deficiencies (C1q, C4, C2) impair clearance of apoptotic debris. DNase I deficiency allows DNA persistence. HLA-DR3, PTPN22 polymorphisms lower the T_reg and B-cell activation thresholds.
• Environmental trigger — UV light induces keratinocyte apoptosis → surface blebs expose nuclear antigens to dermal dendritic cells.
Step 2 — Autoantibody generation
Persistent nuclear-antigen presentation activates autoreactive B cells (with T_H help) → production of multiple anti-nuclear antibodies (ANA):
• Anti-dsDNA — highly specific for SLE; titres correlate with disease activity (especially nephritis).
• Anti-Sm (Smith antigen, small nuclear RNP) — highly specific for SLE (not as sensitive).
• Anti-histone — seen in drug-induced lupus.
• Anti-Ro (SSA) / Anti-La (SSB) — associated with neonatal lupus and Sjögren overlap.
• Anti-phospholipid antibodies — anticardiolipin, lupus anticoagulant → antiphospholipid syndrome (thrombosis, pregnancy loss).
Step 3 — Immune-complex (Type III hypersensitivity) deposition
Autoantibody–antigen complexes form in the circulation and deposit in vessel walls, glomeruli, skin, synovium, and serosal surfaces → activate complement → attract neutrophils → neutrophil extracellular traps (NETs) release more nuclear antigens → self-amplifying cycle.
SLE Pathogenesis