Page 2 of 22
PA8.1-2 | Immunity & Hypersensitivity Reactions — SDL Guide (Part 2)
Type I Hypersensitivity — Immediate/Anaphylactic
Type I is mediated by IgE bound to mast cells (in tissue) and basophils (in blood). It proceeds in two phases:
Phase 1 — Sensitization (silent):
1. First antigen exposure → antigen presentation to Th2 cells
2. Th2 cells release IL-4/IL-13 → B cells undergo IgE class-switching
3. Antigen-specific IgE is secreted and binds the high-affinity Fcε receptor (FcεRI) on mast cells and basophils
4. The individual is now sensitized — no symptoms yet
Phase 2 — Effector (symptomatic):
1. Re-exposure to same antigen → antigen cross-links two adjacent IgE molecules on mast cell surface
2. FcεRI aggregation → mast cell activation → immediate degranulation
Early phase (minutes): Pre-formed mediators released:
• Histamine — vasodilation, increased vascular permeability, bronchospasm, pruritus
• Heparin, proteases (tryptase, chymase)
• Platelet-activating factor (PAF)
• Eosinophil and neutrophil chemotactic factors
Late phase (4–8 hours): Newly synthesised mediators:
• Leukotrienes (LTC4, LTD4, LTE4 = slow-reacting substances of anaphylaxis, SRS-A) — potent bronchospasm
• Prostaglandins (PGD2) — vasodilation, bronchospasm
• Cytokines (IL-4, IL-5, TNF) — sustain eosinophil influx, perpetuate late-phase
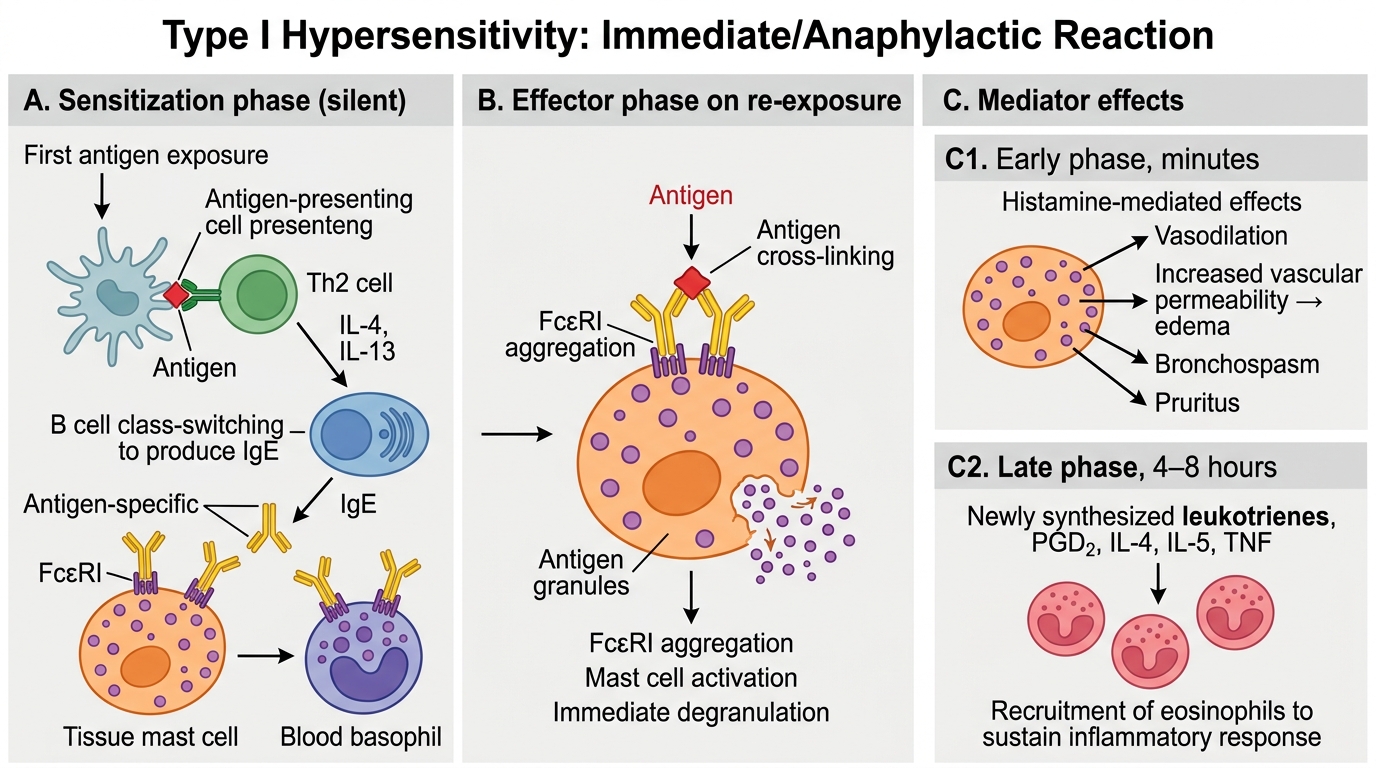
Type I Hypersensitivity: Sensitization and Effector Phases
Clinical spectrum:
• Anaphylaxis — systemic; histamine-mediated hypotension + bronchospasm; life-threatening
• Atopy — familial predisposition to localised Type I reactions (IgE-prone immune bias)
• Bronchial asthma (extrinsic/allergic) — inhaled allergens; bronchospasm via leukotrienes
• Allergic rhinitis — nasal mucosa; sneezing, rhinorrhoea
• Urticaria/Angioedema — skin/subcutaneous; wheal-and-flare
• Food allergy — peanut, shellfish (IgE-mediated; can trigger systemic anaphylaxis)
CLINICAL PEARL
Tryptase as a diagnostic marker of anaphylaxis: Mast cell tryptase is released exclusively during degranulation — it is not present in basophils. A serum tryptase drawn within 1–3 hours of a suspected anaphylactic event and elevated > 11.4 ng/mL strongly supports mast cell activation. Histamine itself clears from serum in < 30 minutes, making it impractical for confirmation. Always send tryptase when anaphylaxis is in the differential, especially for intraoperative reactions where the trigger may be unclear.
Allergy testing rationale: Skin prick testing introduces a tiny amount of allergen into the dermis — if IgE-sensitized mast cells are present, a wheal-and-flare reaction develops within 15 minutes (the Type I effector phase in miniature). A negative skin test reliably excludes IgE-mediated allergy to that specific antigen.
SELF-CHECK
A 30-year-old atopic woman presents with worsening asthma that is poorly controlled despite inhaled corticosteroids. Her physician considers a leukotriene receptor antagonist (montelukast). Which mast cell mediator, released in the late phase of Type I hypersensitivity, is montelukast specifically targeting?
A. Histamine (H1 receptor-mediated bronchoconstriction)
B. Platelet-activating factor (PAF)
C. Leukotrienes LTC4/LTD4/LTE4 (formerly called SRS-A)
D. Tryptase-mediated smooth muscle remodelling
Reveal Answer
Answer: C. Leukotrienes LTC4/LTD4/LTE4 (formerly called SRS-A)
Montelukast blocks the CysLT1 receptor, antagonising the cysteinyl leukotrienes LTC4, LTD4, and LTE4 — the principal late-phase bronchoconstrictors synthesised de novo from arachidonic acid by mast cells and eosinophils via the 5-lipoxygenase pathway. These were historically called the "slow-reacting substances of anaphylaxis (SRS-A)" before their chemical structures were identified. Antihistamines (targeting histamine) are effective for early-phase symptoms (urticaria, rhinorrhoea) but have minimal bronchodilatory effect in chronic asthma.
Type II Hypersensitivity — Antibody-Mediated (Cytotoxic)
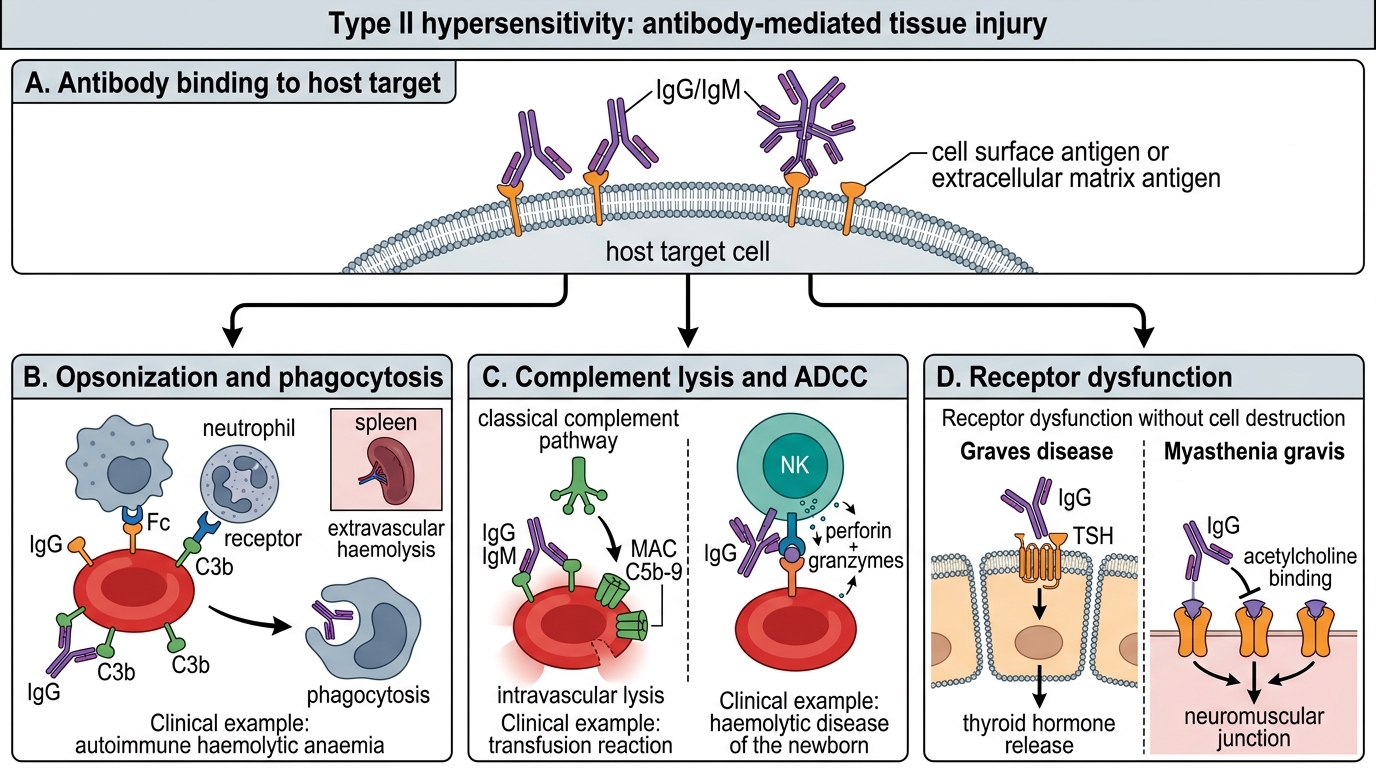
Type II Hypersensitivity: Antibody-Mediated Injury
In Type II reactions, IgG or IgM antibodies bind to antigens displayed on cell surfaces or extracellular matrix and damage host tissues through three distinct mechanisms:
Mechanism 1 — Opsonization and phagocytosis:
• Antibody coats the target cell → Fc receptors on macrophages/neutrophils recognise the Fc tail → phagocytosis and destruction
• Complement activation (C3b deposition) amplifies opsonization
• Example: Autoimmune haemolytic anaemia — IgG against red cell antigens (Rh, Kidd, Duffy) → extravascular haemolysis in spleen
Mechanism 2 — Complement- and NK-cell–mediated lysis (ADCC):
• IgG/IgM binds target → activates classical complement pathway → MAC (C5b-9) punches hole in cell membrane → lysis
• Or: NK cells recognise Fc of bound IgG → release perforin/granzymes → antibody-dependent cellular cytotoxicity (ADCC)
• Example: Transfusion reactions (incompatible blood → intravascular haemolysis); Haemolytic disease of the newborn (HDN) — maternal IgG anti-D crosses placenta
Mechanism 3 — Receptor dysfunction (without cell destruction):
• Antibody binds a receptor and either activates it (agonist) or blocks it (antagonist) → functional disease without major inflammation
• Example (agonist): Graves disease — IgG against TSH receptor mimics TSH → constitutive thyroid stimulation → hyperthyroidism
• Example (antagonist): Myasthenia gravis — IgG against acetylcholine receptor (AChR) at neuromuscular junction → receptor internalisation/blockade → muscle weakness
Other Type II examples:
• Goodpasture syndrome — IgG against type IV collagen in glomerular basement membrane (GBM) and alveolar BM → pulmonary haemorrhage + rapidly progressive GN
• Pemphigus vulgaris — IgG against desmoglein in epidermal desmosomes → acantholysis → skin blisters
• Immune thrombocytopenic purpura (ITP) — IgG against platelet GPIIb/IIIa → platelet destruction
SELF-CHECK
A 25-year-old woman is found to have anti-acetylcholine receptor antibodies. Her symptoms include fatigable ptosis and proximal muscle weakness worsening in the evening. This is BEST classified as which type of hypersensitivity, and through which specific mechanism?
A. Type I — mast cell degranulation at the neuromuscular junction
B. Type II — receptor blockade/internalisation by IgG (functional without lysis)
C. Type III — immune complex deposition in synovial membranes
D. Type IV — CD8⁺ CTL destruction of motor end-plates
Reveal Answer
Answer: B. Type II — receptor blockade/internalisation by IgG (functional without lysis)
Myasthenia gravis is Type II hypersensitivity via the receptor dysfunction mechanism: IgG antibodies bind and cross-link AChR, causing receptor internalisation and complement-mediated receptor degradation, not cell lysis. No immune complexes are deposited (not Type III), and T cells are not the direct effectors (not Type IV). The fatigable pattern results from progressive depletion of functional AChR at the end-plate, worsening with repeated nerve stimulation.
Type III Hypersensitivity — Immune Complex–Mediated
Type III reactions are caused by immune complex (IC) deposition — antigen-antibody (IgG, IgM) complexes that form in circulation or in situ and deposit in vessel walls or tissue, triggering inflammation.
Pathogenesis — four steps:
1. Complex formation: Antigen excess over antibody produces small, soluble complexes that evade phagocytosis; antibody excess produces large, rapidly cleared complexes; slight antigen excess produces the pathological intermediate-sized complexes
2. Deposition: Complexes lodge in capillaries and venules — especially in kidney glomeruli, synovial vessels, skin, and choroid plexus — driven by size, charge, and haemodynamic factors
3. Complement activation: Deposited IC activate classical pathway → C3a/C5a (anaphylatoxins: vasodilation, mast cell degranulation) + C5a (potent neutrophil chemotactant)
4. Neutrophil infiltration and tissue damage: Neutrophils attempt to phagocytose IC but fail (IC are bound to basement membranes) → frustrated phagocytosis → release of lysosomal enzymes and reactive oxygen species → fibrinoid necrosis of vessel walls (vasculitis)
Hallmark histology: Granular/lumpy-bumpy deposits on immunofluorescence (IF) — distinguish from the linear IF pattern of Goodpasture (Type II)
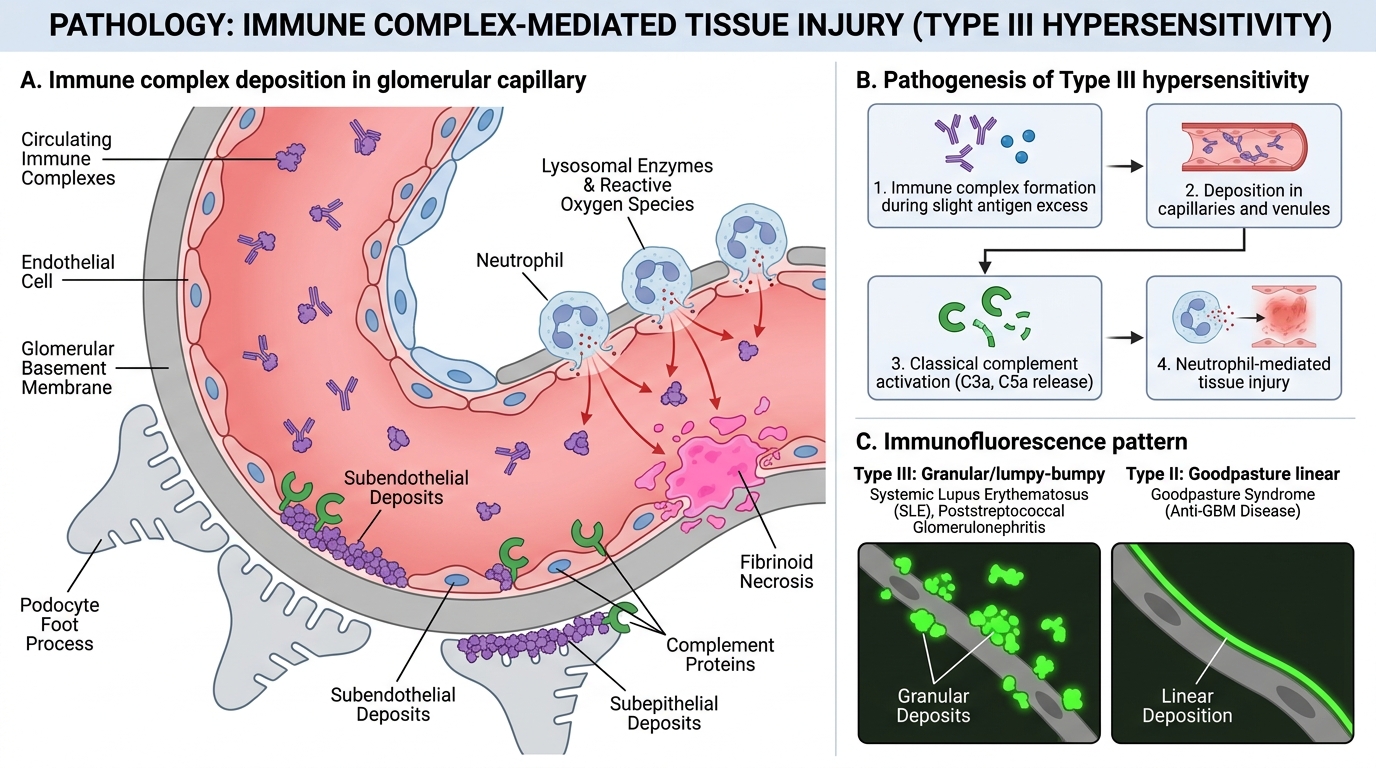
Type III Hypersensitivity: Immune Complex–Mediated Injury
Clinical examples:
| Disease | Antigen | Site of deposition |
|---|---|---|
| Systemic Lupus Erythematosus (SLE) | Nuclear antigens (dsDNA, Sm) | Glomeruli, skin, joints, vessels |
| Post-streptococcal GN | Streptococcal antigens | Glomeruli (subepithelial humps) |
| Serum sickness | Foreign protein (antiserum) | Systemic — joints, skin, kidneys |
| Arthus reaction | Local antigen injection (e.g., vaccine) | Skin/subcutaneous (localised) |
| Infective endocarditis | Bacterial antigens | Glomeruli (focal GN) |
Arthus vs serum sickness: Both are Type III. Arthus is a localised reaction at the site of antigen injection (raised wheals, necrosis at the injection site within 4–8 h); serum sickness is systemic (fever, arthralgia, urticarial rash, lymphadenopathy, GN, 7–10 days after antigen exposure).
CLINICAL PEARL
The complement consumption pattern distinguishes Type II from Type III renal disease: In Goodpasture syndrome (Type II), serum complement levels are normal — antibodies bind directly to the GBM without activating complement in the fluid phase. In SLE nephritis and post-streptococcal GN (Type III), serum C3 and C4 are low (complement is consumed by immune complexes in circulation and glomeruli). Checking serum C3/C4 in a new nephritis case is a rapid, cheap screen to guide further workup — low complement points to Type III (or MPGN Type I), while a normal complement level favours Type II, anti-GBM, or pauci-immune ANCA vasculitis.
SELF-CHECK
A 19-year-old woman develops fever, migratory joint pain, urticarial rash, and mild proteinuria 9 days after receiving anti-snake venom (equine serum). Serum C3 is low. Renal biopsy shows granular IgG deposits on immunofluorescence. This is BEST explained by:
A. Type I — IgE-mediated mast cell degranulation to horse serum proteins
B. Type II — IgG antibodies against glomerular basement membrane components
C. Type III — immune complex deposition; horse serum protein is the antigen
D. Type IV — T-cell–mediated delayed hypersensitivity to equine proteins
Reveal Answer
Answer: C. Type III — immune complex deposition; horse serum protein is the antigen
This is classic serum sickness — a systemic Type III reaction. The foreign horse serum proteins act as antigen; the patient's own IgG antibodies form intermediate-sized immune complexes 7–10 days post-exposure (time for primary IgG production). Complexes deposit systemically (skin, joints, glomeruli), activate complement (hence low C3), and produce neutrophilic vasculitis. Granular IF is the hallmark; linear IF would instead point to Goodpasture (Type II). Type I is excluded by the delay (9 days, not minutes) and requires prior sensitization.