Page 3 of 32
PA24.{1,6} | Bilirubin Metabolism, Jaundice & LFT Interpretation — SDL Guide (Part 3)
Hereditary Hyperbilirubinaemia Syndromes
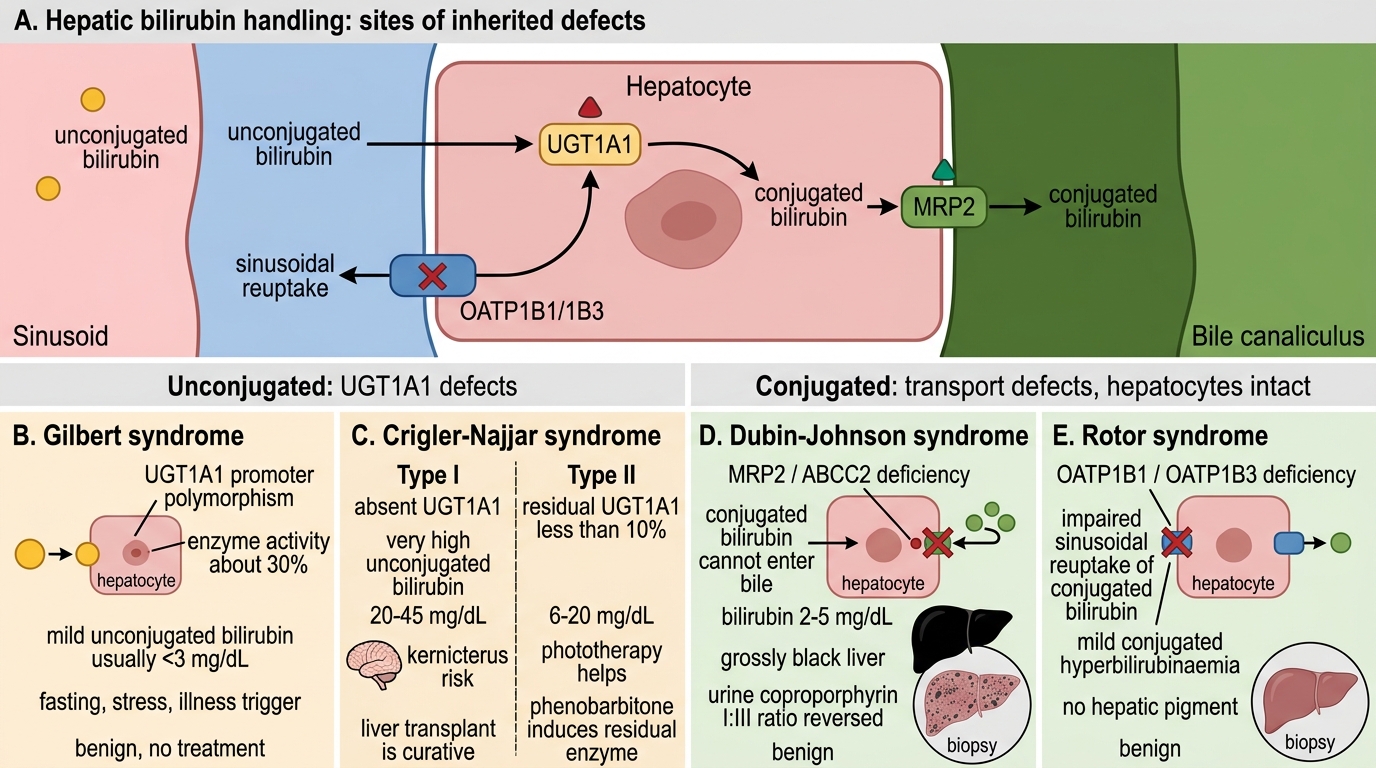
Hereditary Hyperbilirubinaemia Syndromes
Four clinically relevant inherited syndromes disrupt specific steps in hepatic bilirubin handling. They are classified by which fraction accumulates:
Unconjugated hyperbilirubinaemia:
- Gilbert syndrome — Autosomal dominant/recessive (UGT1A1 promoter polymorphism, enzyme activity ≈30%). Mild unconjugated hyperbilirubinaemia (usually <3 mg/dL), precipitated by fasting, stress, intercurrent illness. Benign. Most common hereditary hyperbilirubinaemia (5–8% population). No treatment needed.
2. Crigler-Najjar syndrome — Severe UGT1A1 deficiency:
- Type I: Complete absence of UGT1A1. Bilirubin 20–45 mg/dL. Kernicterus within 18 months without liver transplant (only cure). Autosomal recessive.
- Type II (Arias syndrome): Residual UGT1A1 (<10%). Bilirubin 6–20 mg/dL. Phototherapy helps. Phenobarbitone induces residual UGT1A1.
Conjugated hyperbilirubinaemia (transport defects — hepatocytes intact):
- Dubin-Johnson syndrome — Autosomal recessive. Deficiency of MRP2 (ABCC2) canalicular transporter → CB cannot enter bile. Bilirubin mildly raised (2–5 mg/dL). Liver appears grossly black (melanin-like pigment accumulation — pathognomonic on biopsy). Urine coproporphyrin I:III ratio reversed. Benign course.
- Rotor syndrome — Autosomal recessive. Deficiency of OATP1B1/1B3 (sinusoidal reuptake of conjugated bilirubin). Mild conjugated hyperbilirubinaemia. Liver biopsy: no pigment (distinguishes from Dubin-Johnson). Also benign.
Memory aid: GC (Gilbert, Crigler-Najjar) = Got Conjugation problem → unconjugated ↑. DR (Dubin-Johnson, Rotor) = Dump/Reuptake problem → conjugated ↑.
Neonatal Jaundice and Kernicterus
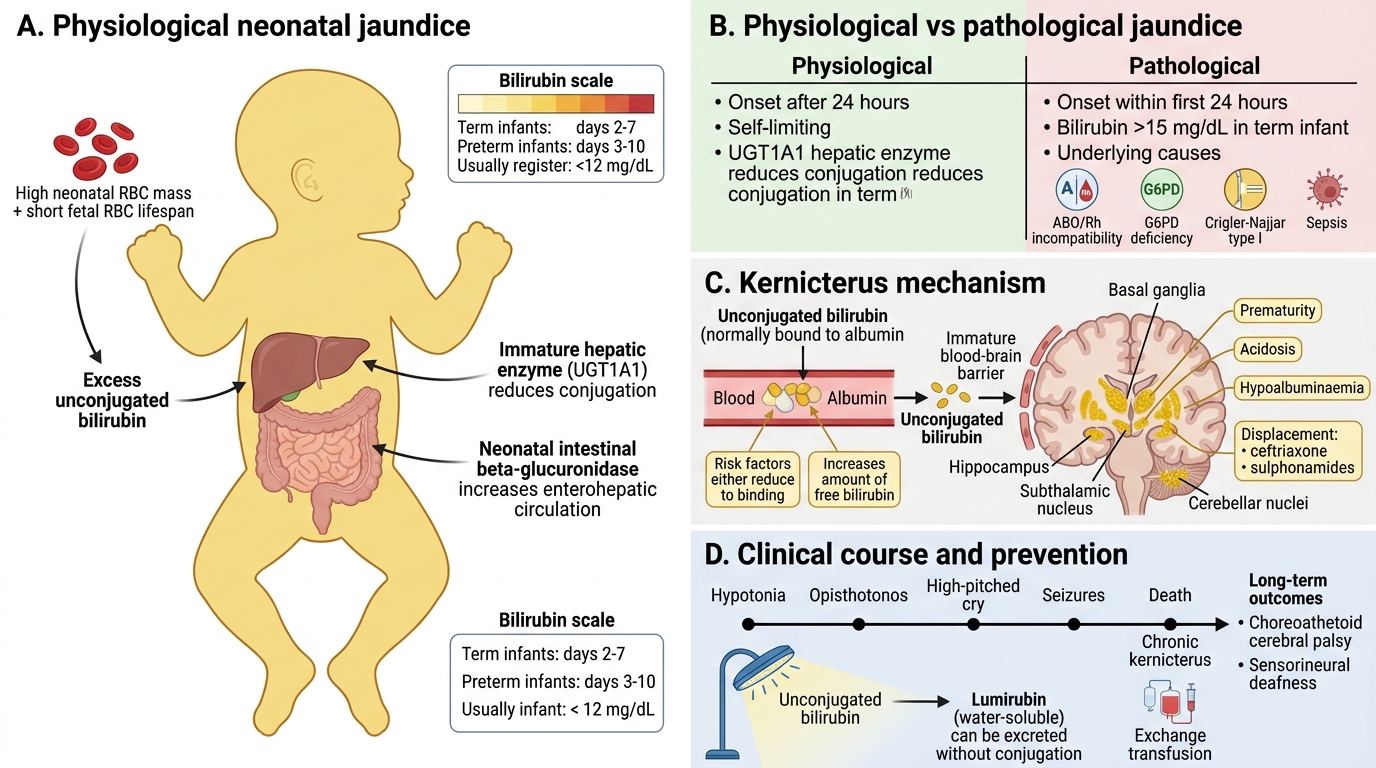
Neonatal Jaundice and Kernicterus
Jaundice affects 60% of term and nearly all preterm neonates in the first week of life. Understanding the mechanism is essential to prevent the irreversible complication of kernicterus (bilirubin encephalopathy).
Physiological neonatal jaundice (days 2–7 in term, days 3–10 in preterm):
- Causes: (a) high RBC mass + short fetal RBC lifespan (70–90 days) → excess UCB production; (b) immature hepatic UGT1A1 (≈1% of adult activity at birth); (c) increased enterohepatic circulation (β-glucuronidase in neonatal gut deconjugates CB → absorbed as UCB).
- Self-limiting: bilirubin rarely exceeds 12 mg/dL in term infants. Resolves as UGT1A1 matures.
Pathological neonatal jaundice (first 24 h, or bilirubin >15 mg/dL term):
- Causes: haemolytic disease of newborn (ABO/Rh incompatibility), G6PD deficiency, Crigler-Najjar Type I, sepsis.
Kernicterus — Unconjugated bilirubin (non-albumin-bound) crosses the immature blood-brain barrier and deposits in the basal ganglia, hippocampus, subthalamic nucleus, and cerebellar nuclei.
- Risk factors: prematurity (lower albumin, immature BBB), acidosis, hypoalbuminaemia, displacement of UCB from albumin by drugs (ceftriaxone, sulphonamides).
- Clinical: hypotonia → opisthotonos → high-pitched cry → seizures → death or choreoathetoid cerebral palsy + sensorineural deafness (chronic Kernicterus).
- Prevention: phototherapy (converts UCB to lumirubin — water-soluble isomer, excreted without conjugation); exchange transfusion for severe cases.
CLINICAL PEARL
Breast milk jaundice (week 2–12) is distinct from breast-feeding jaundice (week 1). Breast milk contains β-glucuronidase and possibly a lipoprotein lipase inhibitor of UGT1A1, causing prolonged unconjugated hyperbilirubinaemia. It is benign and self-resolving, but the mother is often unnecessarily advised to stop breastfeeding — this is incorrect. Temporarily stopping breastfeeding for 24–48 h confirms the diagnosis (bilirubin drops rapidly) but is usually unnecessary.
LFT Interpretation: The Four Enzyme Groups
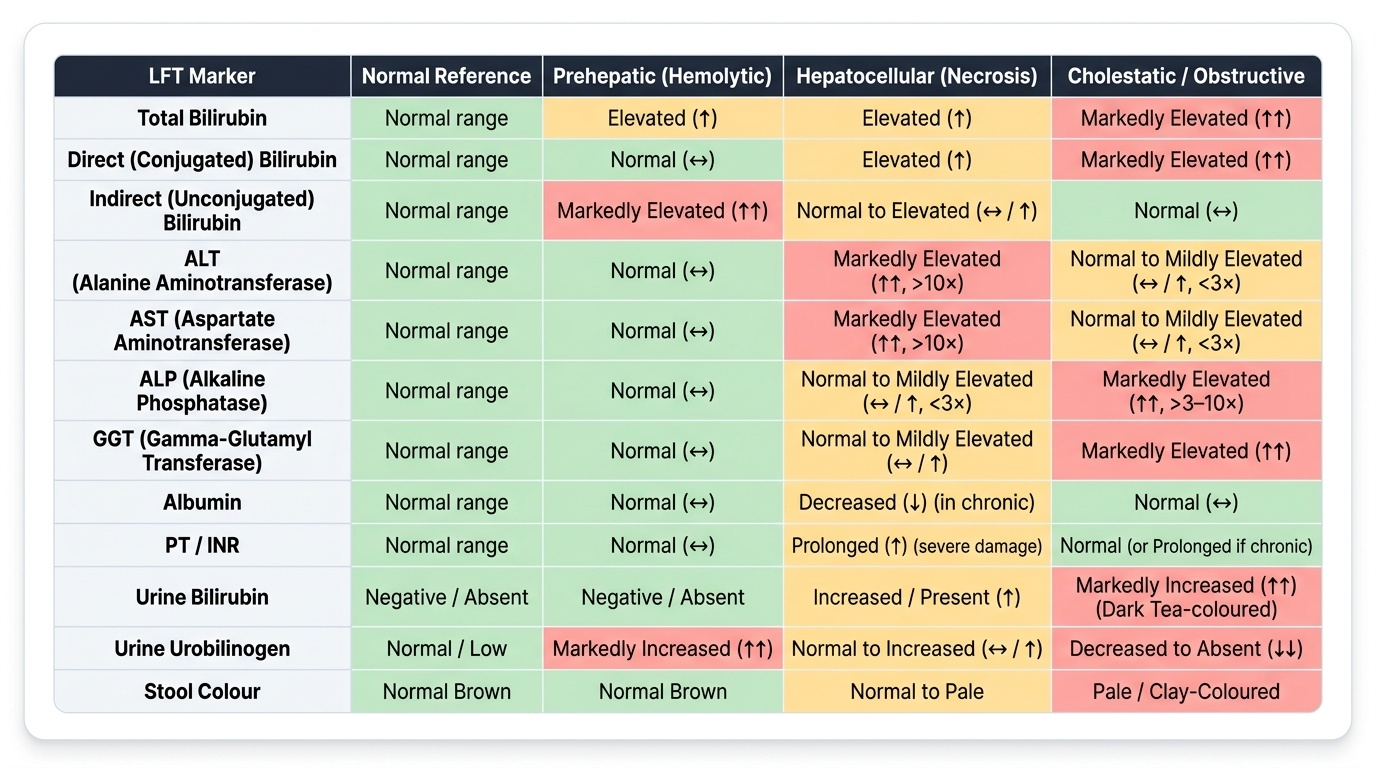
Provided image
A liver function test (LFT) panel contains markers from four functional domains. Learning to read the pattern, not individual values, is the clinical skill:
1. Hepatocellular markers (cytolysis/necrosis):
- Alanine aminotransferase (ALT) — cytosolic, liver-specific. Best marker of hepatocyte injury.
- Aspartate aminotransferase (AST) — cytosolic + mitochondrial; also in cardiac muscle, skeletal muscle, RBCs. Less specific than ALT.
- Elevation: 3–10× = mild hepatitis or fatty liver; >10× = viral hepatitis, toxin, ischaemia; >40× (>1,000 U/L) = fulminant hepatic failure, ischaemic hepatitis ('shock liver'), paracetamol toxicity.
2. Cholestatic markers (biliary):
- Alkaline phosphatase (ALP) — also elevated in bone disease (Paget, fractures, pregnancy). Confirm liver origin with GGT elevation.
- Gamma-glutamyl transferase (GGT) — liver-specific; most sensitive marker of alcohol use and biliary obstruction. Elevated by enzyme-inducing drugs (phenytoin, rifampicin) even without cholestasis.
3. Bilirubin fractions:
- Total, direct (conjugated), indirect (unconjugated) — already covered in previous blocks.
4. Synthetic markers (liver reserve):
- Albumin (t½ = 20 days) — low in chronic liver disease (cirrhosis) and malnutrition; normal in acute hepatitis (takes weeks to fall).
- Prothrombin time (PT/INR) — clotting factors II, V, VII, X, fibrinogen all made in liver; PT rises within 24–48 h of severe hepatocyte loss (useful acute marker). Coagulation also impaired by vitamin K malabsorption in cholestasis (fat-soluble vitamin).
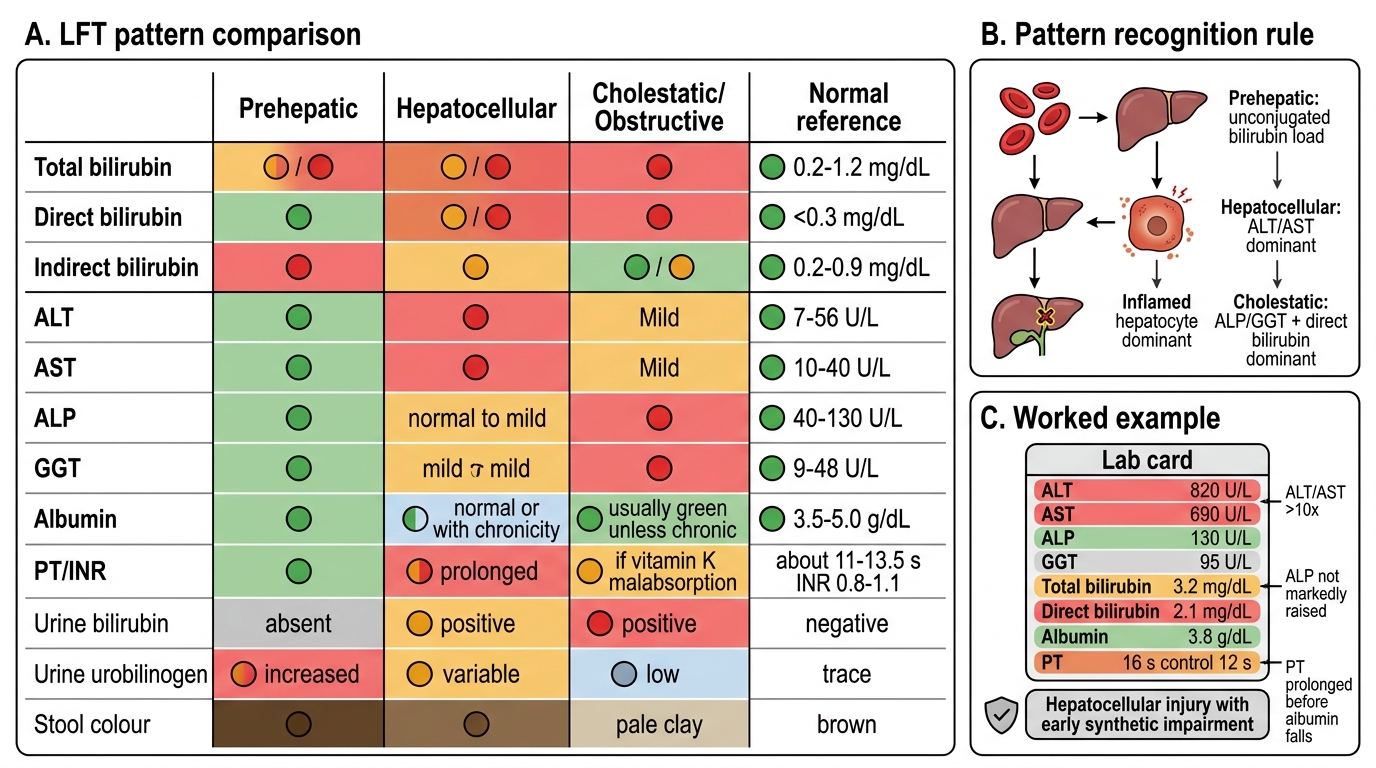
LFT Pattern Recognition: Prehepatic vs Hepatocellular vs Cholestatic
SELF-CHECK
A 45-year-old man is evaluated for elevated liver enzymes. LFT shows: ALT 820 U/L, AST 690 U/L, ALP 130 U/L (normal 40–130), GGT 95 U/L, total bilirubin 3.2 mg/dL (direct 2.1), albumin 3.8 g/dL, PT 16 s (control 12 s). Which pattern does this represent?
A. Hepatocellular injury pattern with early synthetic impairment
B. Isolated prehepatic hyperbilirubinaemia
C. Primary cholestatic disease
D. Physiological neonatal jaundice
Reveal Answer
Answer: A. Hepatocellular injury pattern with early synthetic impairment
Markedly elevated transaminases (ALT > AST, both >10×) with disproportionately normal ALP and GGT is the classic hepatocellular pattern — viral hepatitis is the leading diagnosis here. Mixed hyperbilirubinaemia (both fractions elevated) and prolonged PT indicate early synthetic impairment (factor production failing). Albumin is still normal (its 20-day half-life means it has not fallen yet). ALP and GGT are at or near the upper limit but not markedly elevated — ruling out primary cholestatic disease.