Page 15 of 32
PA24.{5,7} | Portal Hypertension & Hepatocellular Carcinoma — SDL Guide (Part 2)
Splenomegaly and Hypersplenism
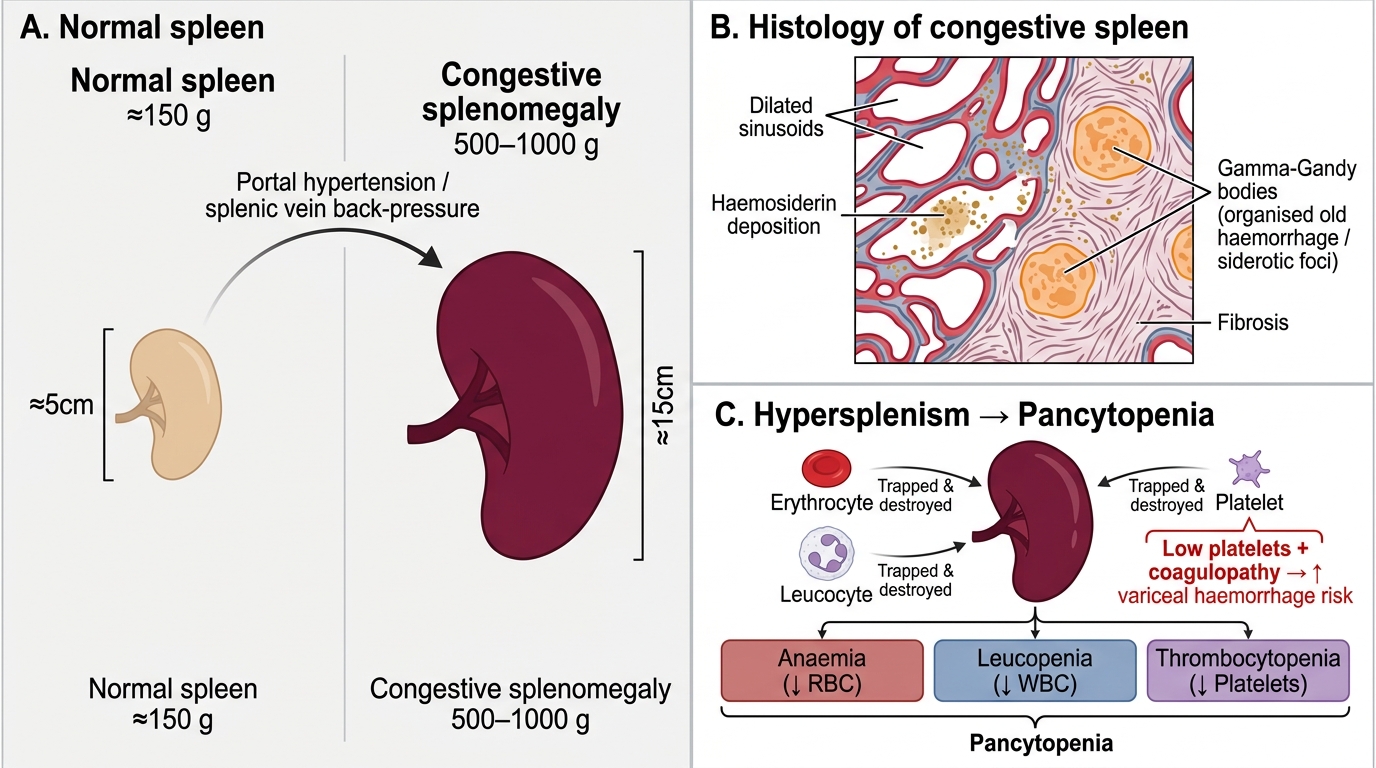
Congestive Splenomegaly and Hypersplenism in Portal Hypertension
Sustained back-pressure in the splenic vein causes congestive splenomegaly. In portal hypertension, the spleen may reach 500–1,000 g (normal ≈ 150 g). Histologically: dilated sinusoids, haemosiderin deposition, fibrosis (Gamna-Gandy bodies = foci of organised old haemorrhage).
Hypersplenism is the clinical consequence: the enlarged spleen traps and destroys circulating blood cells excessively, causing pancytopenia — anaemia, leucopenia, and thrombocytopenia. A low platelet count in a cirrhotic patient almost always reflects hypersplenism. Paradoxically, thrombocytopenia combined with coagulopathy (reduced hepatic synthesis of clotting factors) severely worsens the risk of variceal haemorrhage.
Ascites — Mechanism
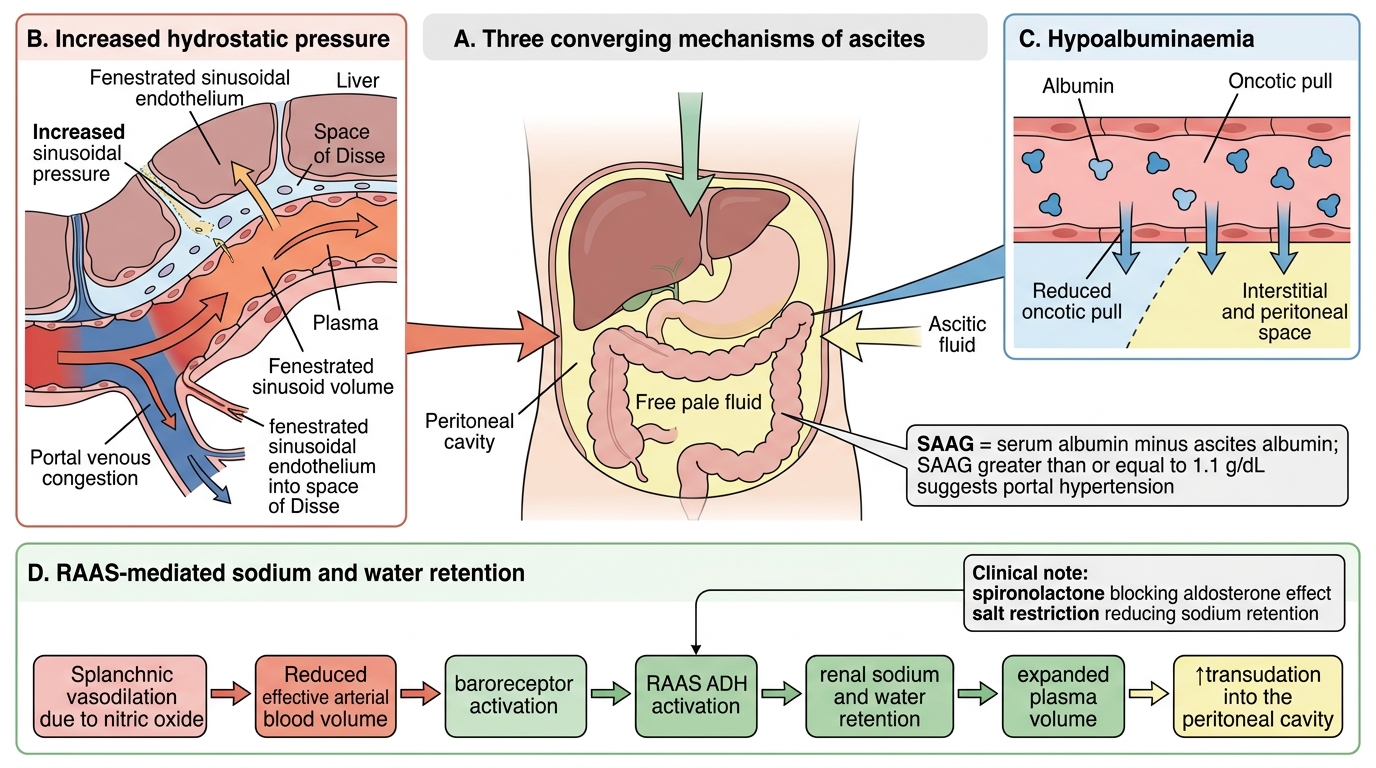
Mechanisms of Ascites in Portal Hypertension
Ascites (free fluid in the peritoneal cavity) in portal hypertension is multifactorial and involves at least three reinforcing mechanisms.
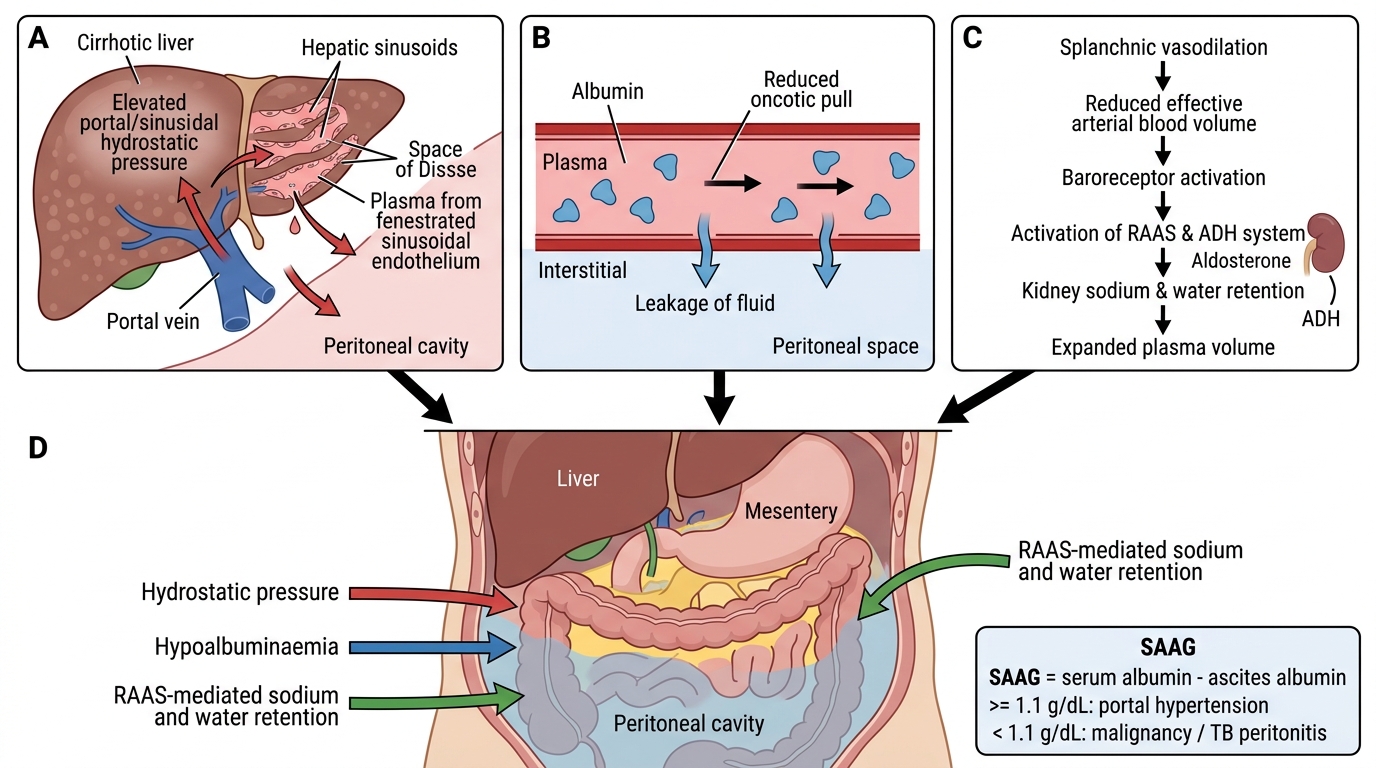
Pathogenesis of Ascites in Portal Hypertension
Mechanism 1 — Increased hydrostatic pressure: Raised sinusoidal pressure drives plasma across the fenestrated sinusoidal endothelium into the space of Disse and then into the peritoneal cavity via the liver surface and mesenteric capillaries. This is the primary driver in early/moderate portal hypertension.
Mechanism 2 — Hypoalbuminaemia: The damaged liver synthesises less albumin. Reduced plasma oncotic pressure favours fluid movement out of vessels. This amplifies fluid accumulation.
Mechanism 3 — RAAS and sodium/water retention: Splanchnic vasodilation (NO-mediated) reduces effective arterial blood volume → baroreceptors activate RAAS and ADH → renal sodium and water retention → expands plasma volume → more fluid transudation into peritoneum. This peripheral vasodilation theory explains why diuretics (spironolactone targets aldosterone) and salt restriction are therapeutic.
Serum-Ascites Albumin Gradient (SAAG): SAAG = serum albumin − ascites albumin. SAAG ≥ 1.1 g/dL indicates portal hypertension as the cause (high specificity). SAAG < 1.1 g/dL suggests exudate causes (malignancy, TB peritonitis).
CLINICAL PEARL
SAAG in daily practice: In Indian hospitals, where both TB peritonitis and cirrhotic ascites are common, SAAG is your first discriminating test — cheap, reliable, calculated from two albumin levels. A SAAG ≥ 1.1 g/dL confidently points to portal hypertension. Do not miss the coexistence of TB + cirrhosis (SAAG may be intermediate 0.8–1.1 g/dL in these cases — always send ascitic fluid ADA and CBNAAT simultaneously).
Hepatic Encephalopathy
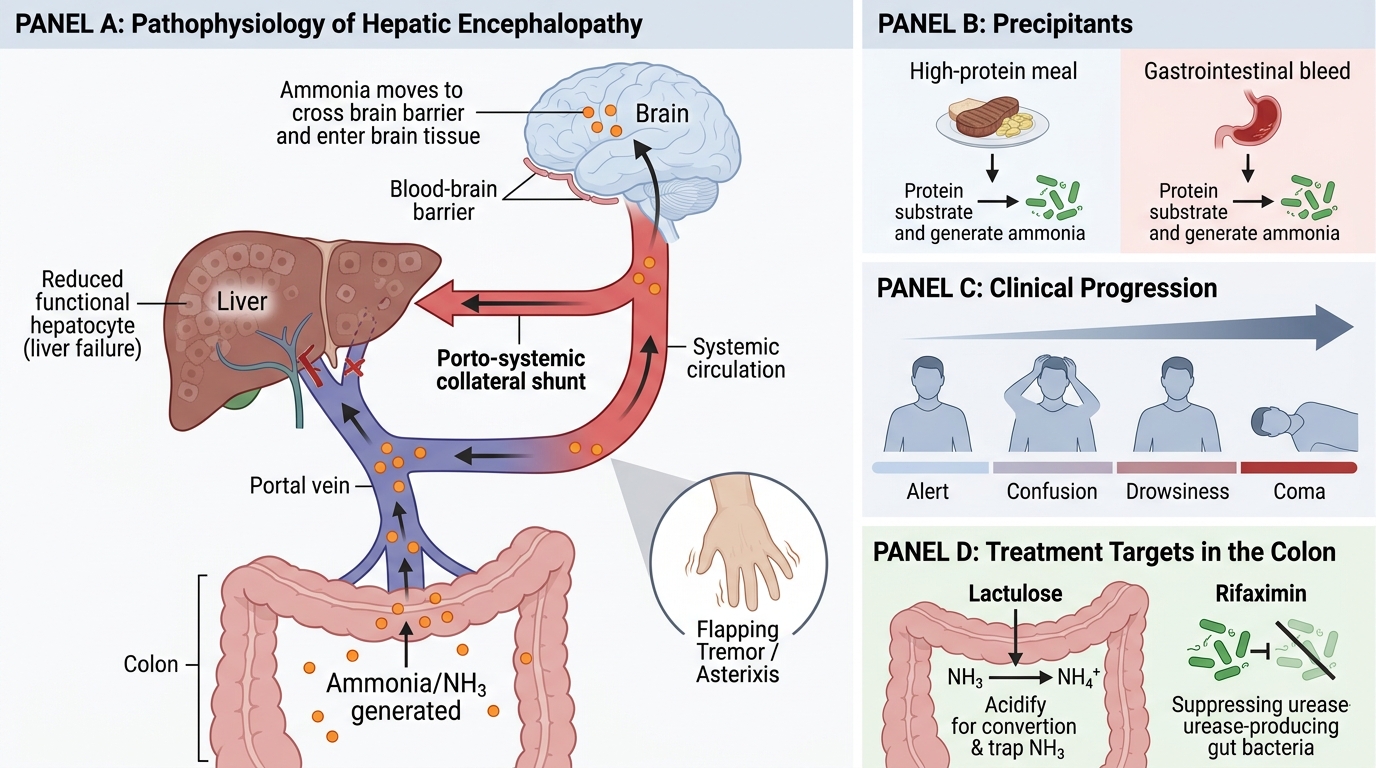
Hepatic Encephalopathy: Ammonia Pathway and Treatment Targets
Hepatic encephalopathy (HE) is a neuropsychiatric syndrome caused by the liver's failure to metabolise gut-derived nitrogenous toxins — principally ammonia — that then enter the systemic circulation and cross the blood-brain barrier.
In portal hypertension, two factors converge:
1. Porto-systemic shunting — blood from the gut bypasses the liver through collaterals, delivering unmetabolised ammonia and other toxins directly to the systemic circulation.
2. Reduced hepatocellular mass — even blood that does pass through the liver encounters insufficient functioning hepatocytes to clear toxins.
Ammonia is converted by gut bacteria from dietary protein. This is why a high-protein meal or a GI bleed (where blood proteins become substrate for colonic bacteria) precipitates acute HE. Clinically: flapping tremor (asterixis), confusion, drowsiness, coma. Treatment targets gut ammonia: lactulose (acidifies colon, traps NH4+), rifaximin (suppresses urease-producing bacteria).