Page 7 of 32
PA24.3 | Viral & Toxic Hepatitis — SDL Guide (Part 2)
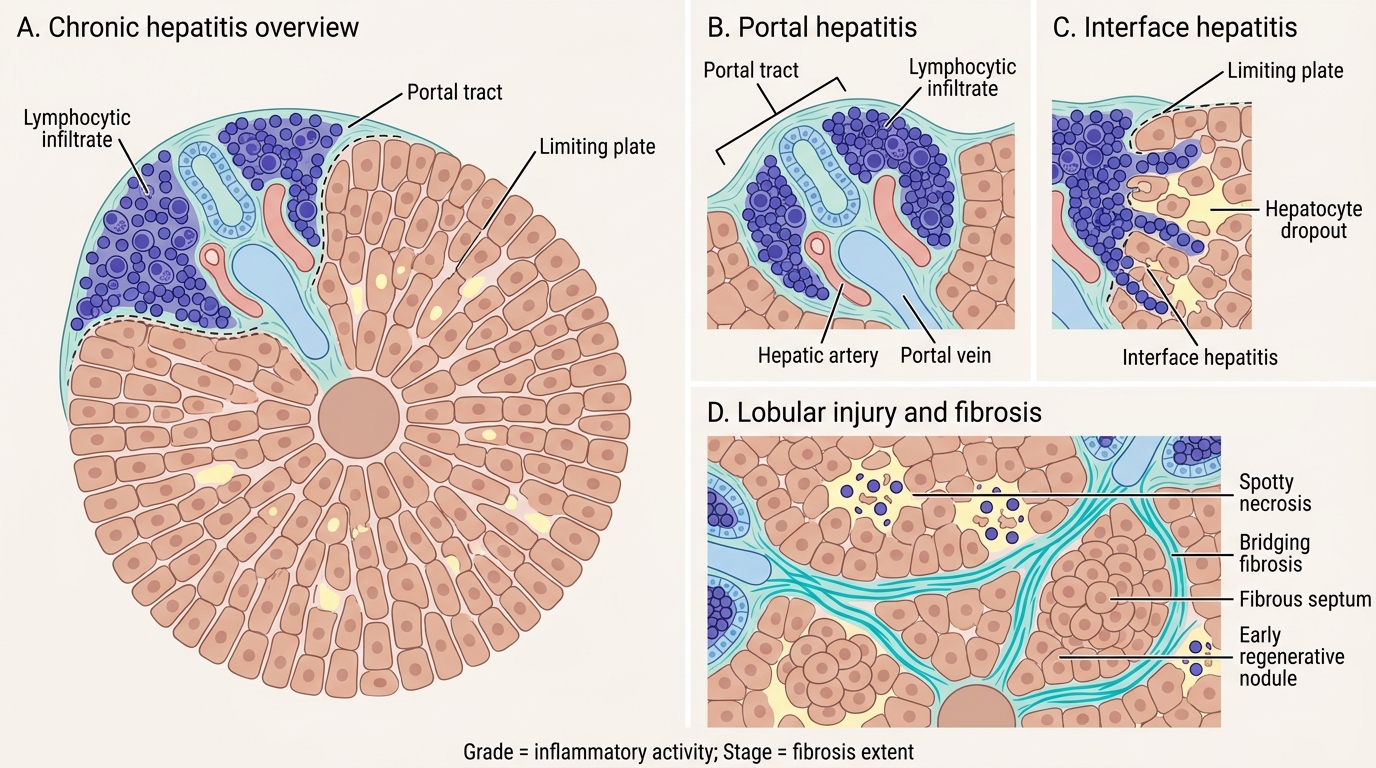
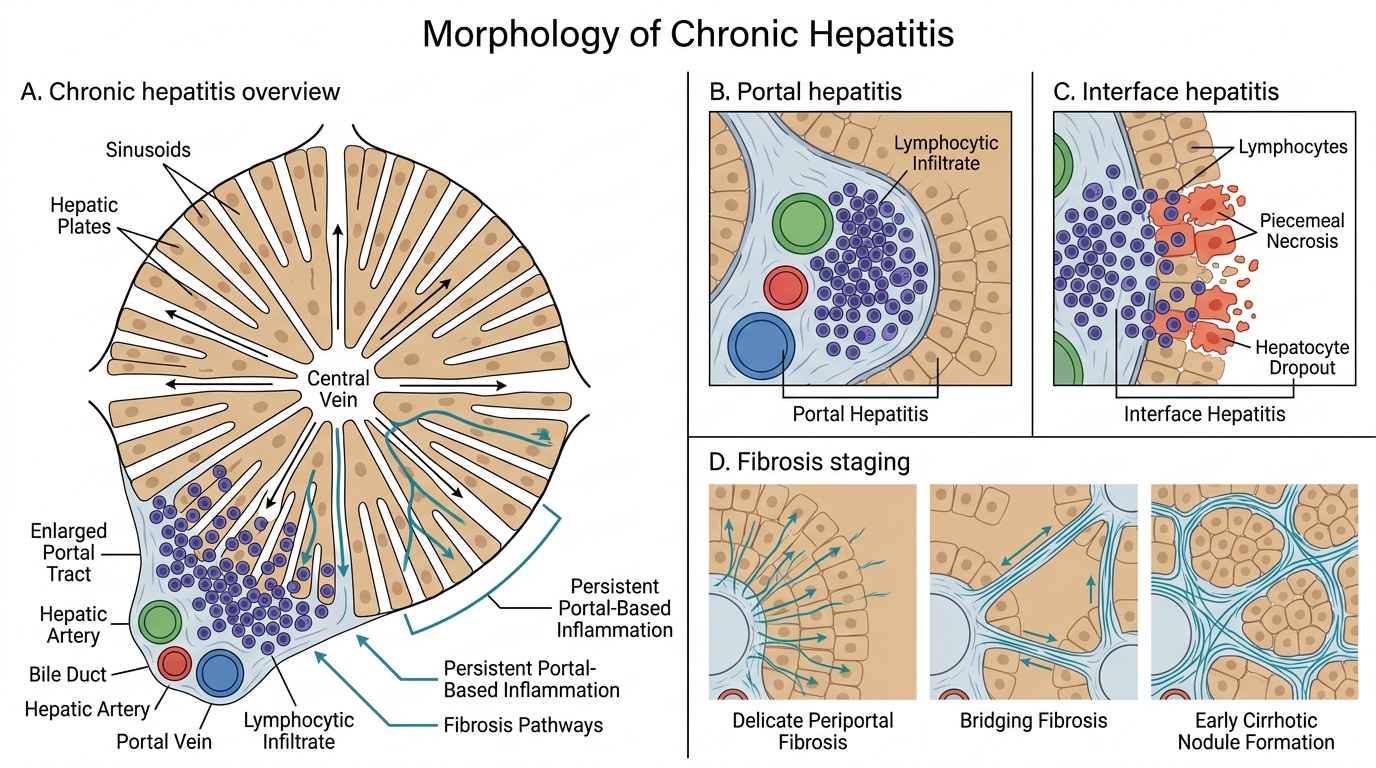
Morphology of Chronic Hepatitis
Morphology of Chronic Hepatitis
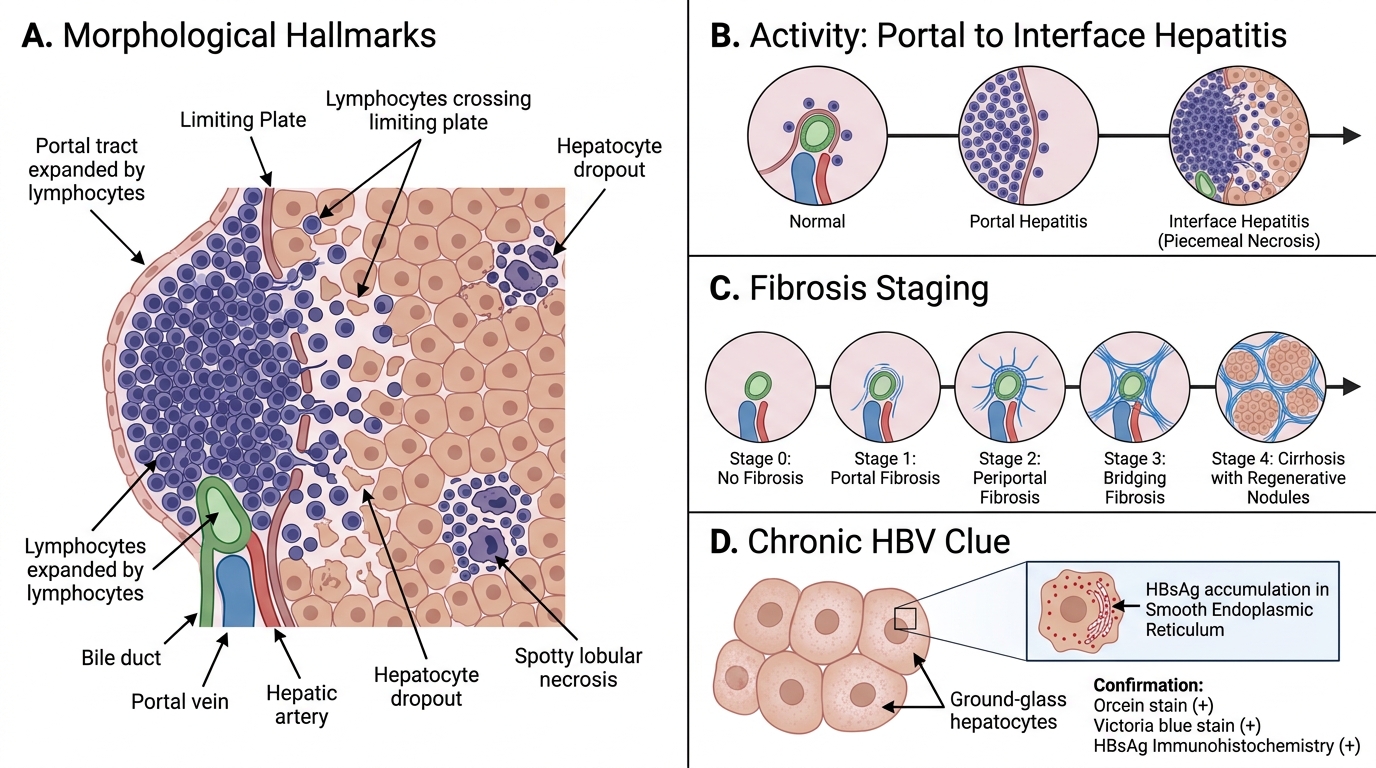
Chronic hepatitis is defined as persistent hepatic inflammation for >6 months. The morphological hallmarks differ from acute hepatitis and are critical for grading (activity) and staging (fibrosis).
Key features:
- Portal hepatitis — lymphocytic infiltrate expanding portal tracts; the mildest form.
- Interface hepatitis (piecemeal necrosis) — lymphocytes spill out of portal tracts and erode the limiting plate (the hepatocyte layer at the portal–lobular interface), causing hepatocyte dropout. This is the defining feature of active chronic hepatitis — the higher the grade, the more interface hepatitis.
- Lobular hepatitis — spotty necrosis within lobules persisting in chronic disease.
- Fibrosis and staging:
- Stage 0 = no fibrosis
- Stage 1 = portal fibrosis (fibrous expansion of portal tracts)
- Stage 2 = periportal fibrosis (fibrous septa beginning to form)
- Stage 3 = bridging fibrosis (portal-portal or portal-central fibrous bridges)
- Stage 4 = cirrhosis (regenerative nodules + diffuse fibrosis)
HBV-specific finding — Ground-glass hepatocytes:
• Hepatocytes with pale, finely granular, eosinophilic cytoplasm filling the cell; seen in chronic HBV.
• Represent massive accumulation of HBsAg in smooth ER (confirmed by orcein stain / Victoria blue stain, or immunohistochemistry for HBsAg).
• Not seen in HCV or other hepatitides — this feature strongly suggests chronic HBV.
Grading (activity: 0–3 or 0–4 on Ishak/Metavir scales) + Staging (fibrosis 0–4) together guide antiviral therapy decisions.
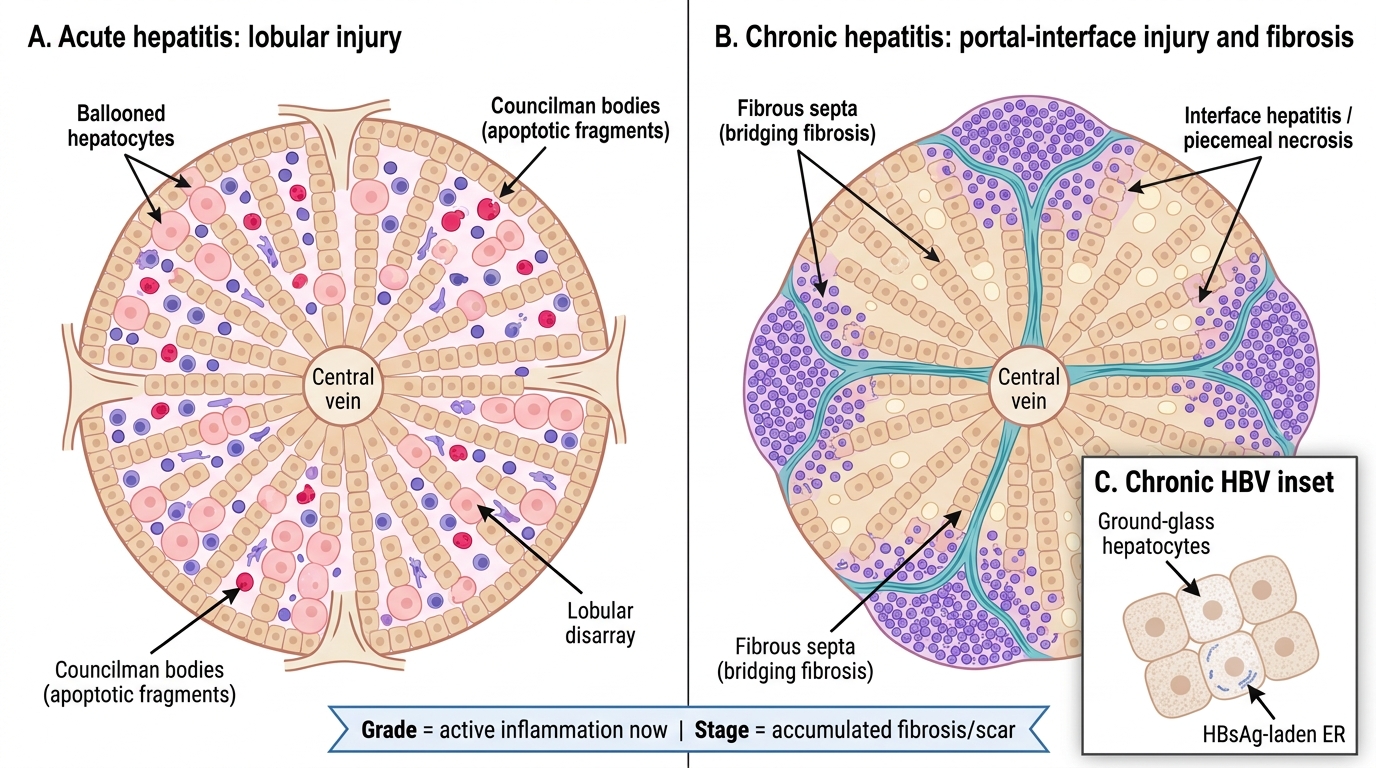
Acute Versus Chronic Hepatitis: Histological Patterns
CLINICAL PEARL
Grading vs Staging — the clinical shortcut: Grade = how active the inflammation is RIGHT NOW (reversible with treatment). Stage = how much scar (fibrosis) has accumulated (largely irreversible). A patient can have high grade but low stage — treat aggressively and you can prevent progression. A stage 3 patient with low grade inflammation still needs antivirals to prevent progression to cirrhosis. Biopsy is being replaced by non-invasive fibroscan/elastography in many centres, but you need to know the histological basis to interpret elastography reports.
Clinical Phases, Outcomes & Complications
Morphology of Chronic Hepatitis
Acute viral hepatitis follows a stereotyped clinical course with four phases:
1. Incubation phase — virus replicates; patient asymptomatic; serum transaminases begin to rise.
2. Pre-icteric (prodromal) phase — flu-like illness: fever, malaise, anorexia, nausea, RUQ discomfort; transaminases peak (can exceed 1000 U/L); viremia at maximum; most infectious.
3. Icteric phase — jaundice appears; symptoms may paradoxically improve as viral load falls (immune response kicking in); dark urine (bilirubinuria), pale stools, pruritus.
4. Recovery phase — jaundice resolves; transaminases normalise over weeks.
Outcomes — the fork in the road:
| Outcome | Key feature |
|---|---|
| Complete resolution | ~95% of HBV adults; all HAV & HEV |
| Carrier state | HBV: HBsAg positive >6 months, normal LFTs, minimal histology |
| Chronic hepatitis | Persistent inflammation >6 months; grades into cirrhosis over years |
| Fulminant hepatic failure | Encephalopathy + coagulopathy within 8 weeks; massive necrosis; <1% of hepatitis but 80% mortality without transplant |
| Cirrhosis | End-stage fibrosis; portal hypertension, liver failure |
| Hepatocellular carcinoma (HCC) | Chronic HBV + cirrhosis: 100-fold increased risk; HCV + cirrhosis: 15–20% lifetime risk |
The carrier state in HBV: HBsAg persists, viral DNA replicates at low levels, no significant hepatitis histologically. BUT: DNA can integrate into the host genome → HCC can develop even WITHOUT cirrhosis in HBV carriers (unlike other causes of HCC). This is why HBsAg-positive carriers need HCC surveillance (AFP + ultrasound every 6 months).
SELF-CHECK
A 55-year-old man has had HBsAg detected for 22 years. His transaminases have been normal throughout. He has never had jaundice. A recent ultrasound shows a 2 cm hepatic nodule with arterial enhancement on CT — suspicious for HCC. Which property of HBV best explains why this carrier developed HCC without cirrhosis?
A. HBV RNA integrates randomly into host chromosomes disrupting tumour suppressor genes
B. HBV DNA can integrate into the host genome, disrupting tumour suppressor genes and activating oncogenes
C. Chronic inflammation from HBV CTL activity drives cumulative mutagenesis
D. HBsAg directly activates the PI3K–mTOR oncogenic pathway in hepatocytes
Reveal Answer
Answer: B. HBV DNA can integrate into the host genome, disrupting tumour suppressor genes and activating oncogenes
HBV has a DNA genome (partially double-stranded) that can integrate into the host genome via its replication cycle (unlike retroviruses, integration is not obligate but occurs frequently). This insertional mutagenesis can disrupt tumour suppressor genes (e.g., TP53) or activate proto-oncogenes near insertion sites — a direct oncogenic mechanism independent of cirrhosis. Option A is wrong because HBV is a DNA virus (not RNA). Option C describes inflammation-driven mutagenesis, which is the HCV/cirrhosis pathway. Option D is not an established primary mechanism. This is why HBV carriers (even without cirrhosis) need HCC surveillance.
Toxic & Drug-Induced Hepatitis — DILI
Morphology of Chronic Hepatitis
Drug-induced liver injury (DILI) is the most common cause of acute liver failure in developed countries and a significant cause in India (largely paracetamol overdose and anti-TB drugs). Two fundamentally different mechanisms:
### Predictable (Dose-Dependent, Intrinsic) DILI
- Occurs in anyone who takes a sufficient dose.
- Reproducible in animal models.
- Latency is short and dose-related.
- Classic example: Paracetamol (acetaminophen) overdose
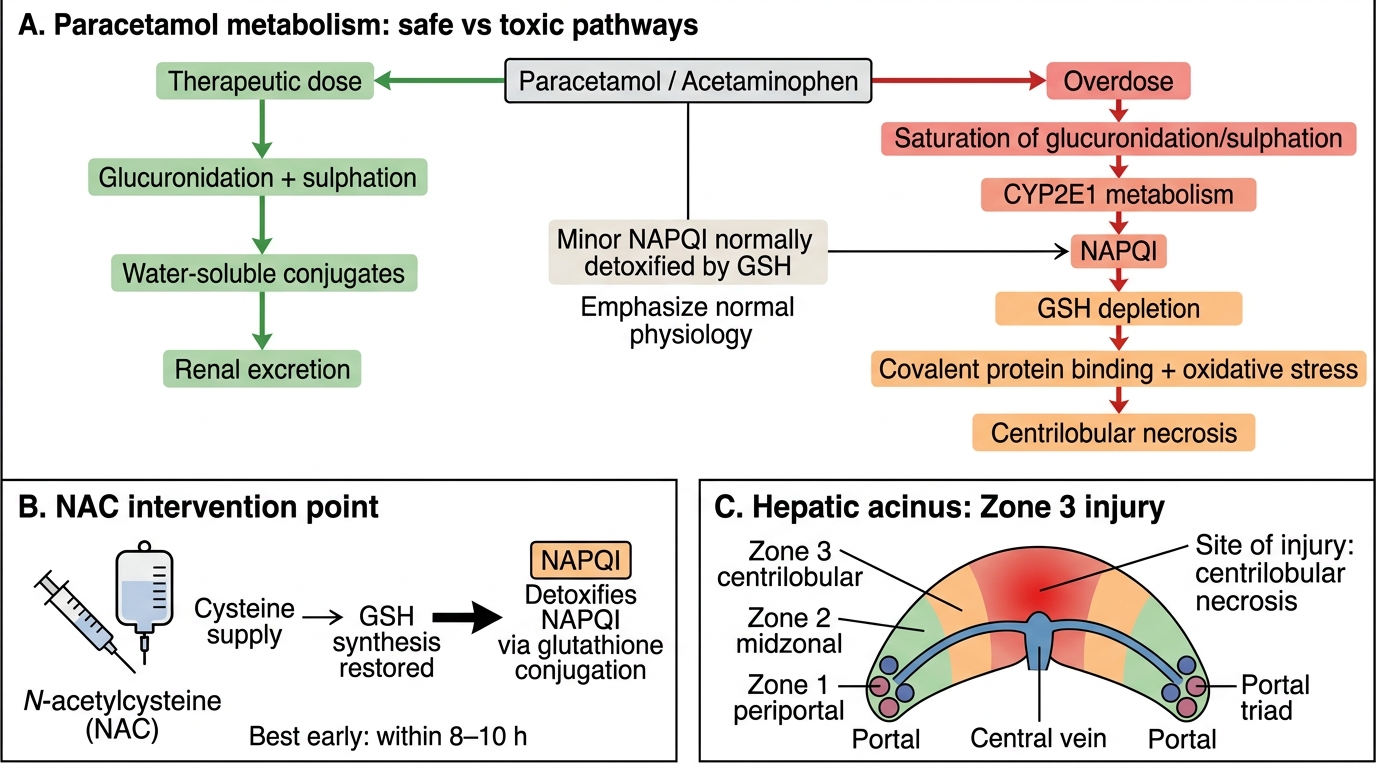
Paracetamol toxicity mechanism:
1. At therapeutic doses: paracetamol conjugated by glucuronidation (UGT) and sulphation → safe, excreted.
2. Small fraction metabolised by CYP2E1 (cytochrome P450) → NAPQI (N-acetyl-p-benzoquinone imine), a highly reactive electrophile.
3. NAPQI rapidly inactivated by conjugation with glutathione (GSH) in hepatocytes.
4. In overdose: glucuronidation/sulphation saturated → all excess paracetamol goes via CYP2E1 → NAPQI overwhelms GSH stores.
5. Free NAPQI binds covalently to hepatocyte proteins → centrilobular (zone 3) necrosis (CYP2E1 is richest in zone 3).
6. Treatment: N-acetylcysteine (NAC) replenishes GSH.
Other predictable hepatotoxins: CCl₄ (carbon tetrachloride) — converted by CYP2E1 to CCl₃• free radical → lipid peroxidation → centrilobular necrosis.
### Idiosyncratic DILI
- Affects only a small minority of exposed individuals (<1 in 1,000–10,000).
- Not reproducible in animal models; unpredictable.
- Two subtypes:
- Metabolic idiosyncrasy — aberrant drug metabolism producing toxic metabolites in genetically susceptible individuals.
- Immune idiosyncrasy — drug or metabolite acts as hapten; triggers autoimmune-like hepatitis (e.g., halothane hepatitis, isoniazid).
Cholestatic vs Hepatocellular Drug Injury
| Pattern | Dominantly elevated | Examples |
|---|---|---|
| Hepatocellular | ALT/AST (>5× ULN) | Paracetamol, isoniazid, statins, ketoconazole |
| Cholestatic | ALP/GGT (>2× ULN) | Chlorpromazine, anabolic steroids, amoxicillin-clavulanate |
| Mixed | Both elevated | Co-amoxiclav, carbamazepine |
Paracetamol Hepatotoxicity Mechanism
SELF-CHECK
A 19-year-old girl is brought in after taking 30 paracetamol tablets 8 hours ago. Her liver enzymes are currently normal but her paracetamol level is dangerously high. The emergency physician starts N-acetylcysteine immediately. What is the principal biochemical rationale for NAC in this setting?
A. NAC inhibits CYP2E1, preventing further NAPQI generation
B. NAC directly neutralises NAPQI by covalent binding
C. NAC replenishes glutathione stores, allowing continued NAPQI detoxification
D. NAC induces UGT enzymes to increase glucuronidation of remaining paracetamol
Reveal Answer
Answer: C. NAC replenishes glutathione stores, allowing continued NAPQI detoxification
NAC is a cysteine pro-drug: it is taken up by hepatocytes and converted to L-cysteine, the rate-limiting substrate for glutathione (GSH) synthesis. By replenishing intracellular GSH, NAC restores the capacity to conjugate and detoxify NAPQI before it binds covalently to cellular proteins. The enzymes are not normal NAC targets (A, D), and NAC does not bind NAPQI directly (B) — its mechanism is indirect via GSH repletion. This is why timing matters: NAC is most effective <8–10 hours post-ingestion, before GSH is irreversibly depleted.