Page 17 of 25
PA27.3-4 | Acute & Chronic Renal Failure — SDL Guide (Part 3)
Pathology of CKD: Gross and Microscopic Features
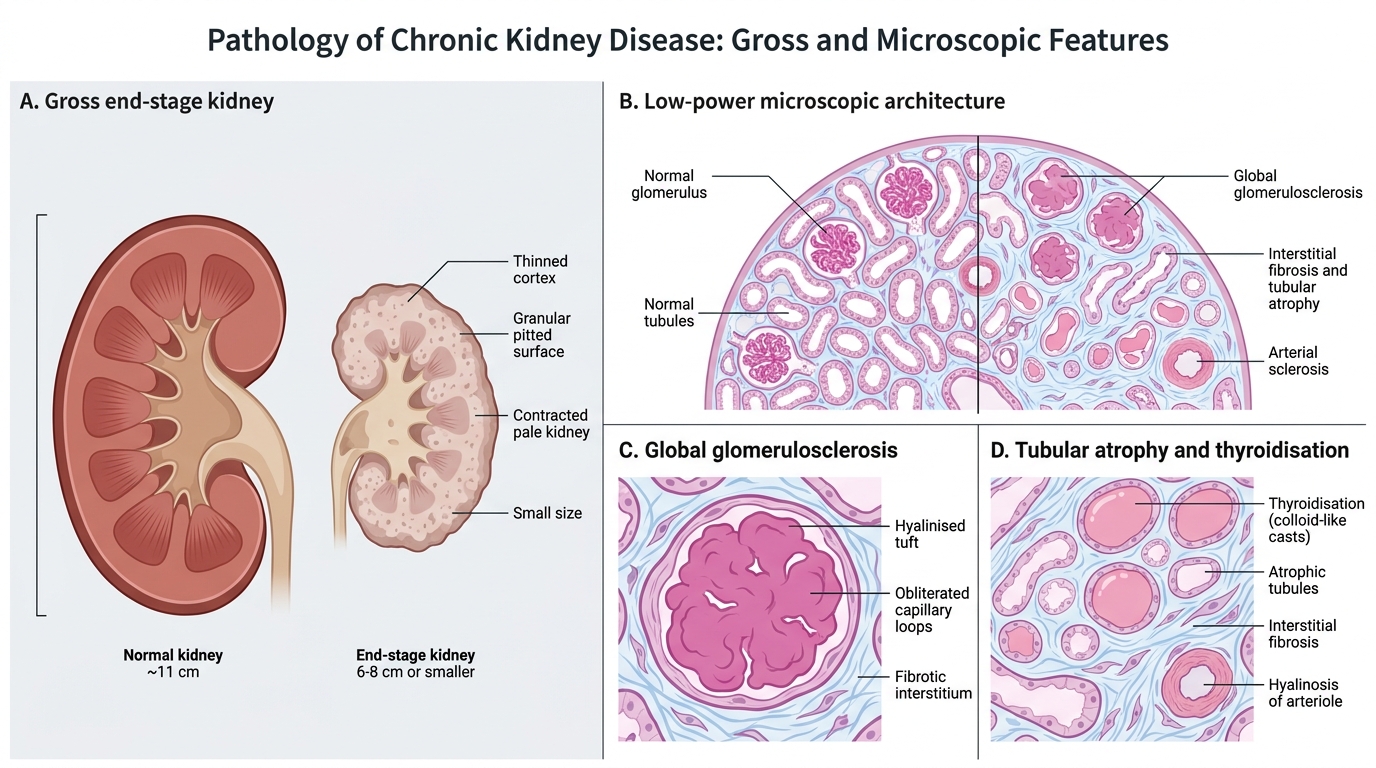
Gross and Microscopic Pathology of Chronic Kidney Disease
The progressive structural changes in CKD converge on a common end-point regardless of the initial cause:
Gross pathology — end-stage kidney:
- Bilaterally small, contracted kidneys (normal: ~11 cm; end-stage: 6–8 cm or smaller)
- Granular/pitted surface — due to scarred nephrons creating irregular depressions between surviving foci
- Thinned cortex (normal: ~1 cm; end-stage: <0.5 cm)
- Firm, fibrous texture; pallor
- Exception: polycystic kidneys are enlarged, not small; diabetic nephropathy kidneys may remain normal size until late.
Microscopic pathology:
- Glomerulosclerosis — global (all capillary loops) or segmental; hyalinisation; obliteration of glomerular tufts
- Tubular atrophy — tubular diameter shrinks; thyroidisation pattern (colloid-like casts in simplified tubules, mimicking thyroid follicles)
- Interstitial fibrosis — collagen deposition replacing lost nephrons (TGF-β driven)
- Arterial and arteriolar hyalinosis/sclerosis — especially in hypertensive and diabetic CKD
- Compensatory hypertrophy — surviving nephrons enlarge (hyperfiltration)
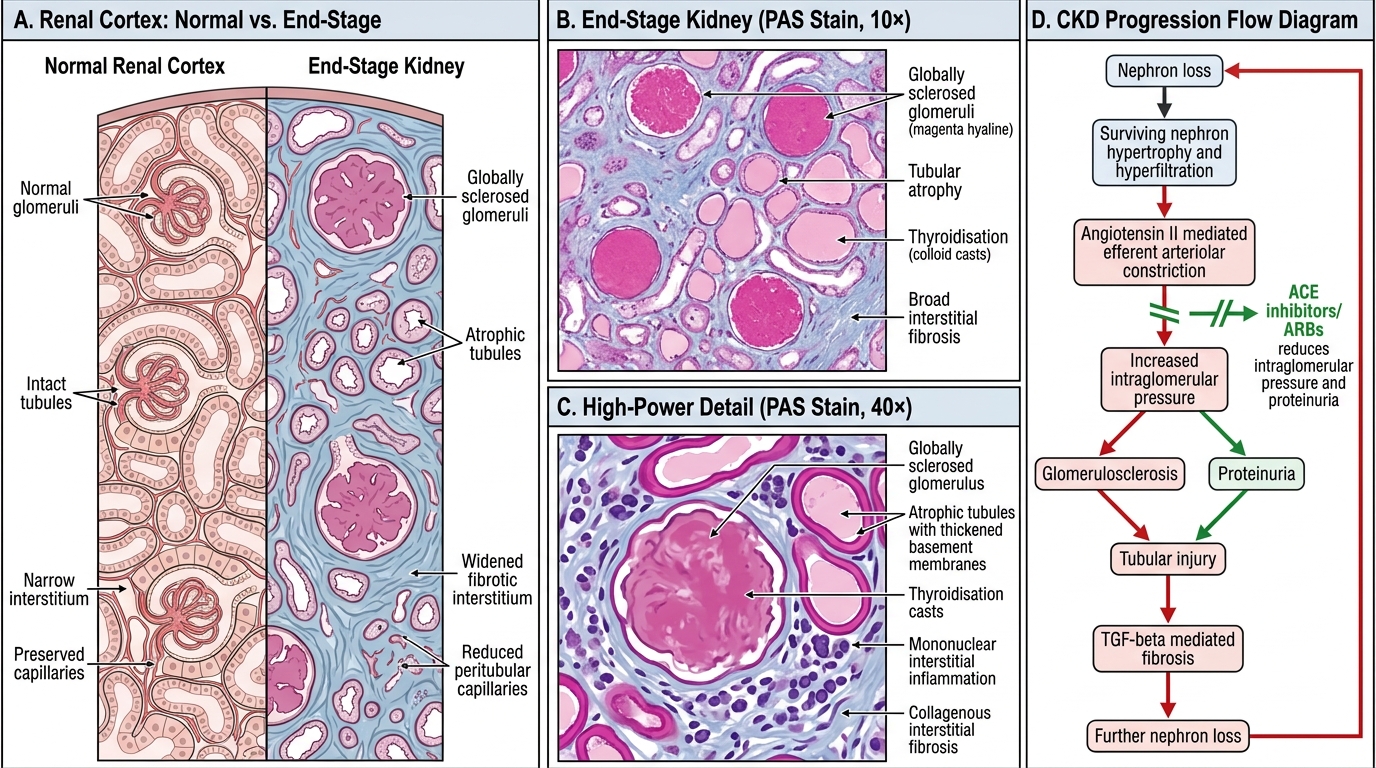
End-Stage Kidney and CKD Progression
Pathophysiology of CKD Progression — Hyperfiltration and RAAS
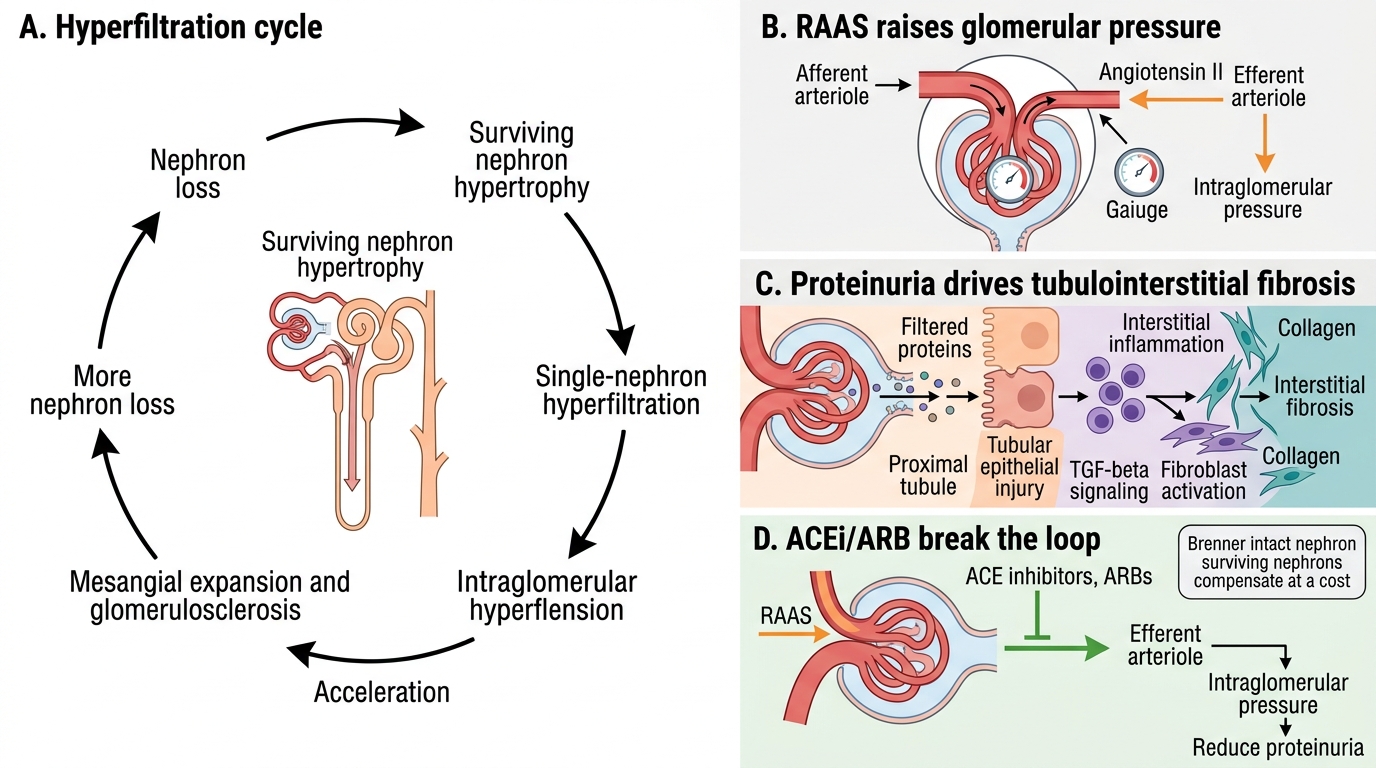
CKD Progression: Hyperfiltration and RAAS
CKD is progressive — even after the initial insult is removed — because of a self-amplifying cycle driven by the surviving nephrons.
The hyperfiltration–proteinuria–fibrosis cycle:
1. Nephron loss → remaining nephrons hypertrophy and hyperfiltrate (↑single-nephron GFR)
2. Hyperfiltration → ↑intraglomerular pressure (efferent arteriolar constriction by angiotensin II)
3. ↑Pressure → glomerular hypertension → mesangial expansion → glomerulosclerosis of remaining nephrons
4. Glomerulosclerosis → more nephron loss → cycle accelerates
5. Simultaneously: proteinuria (filtered proteins are directly toxic to tubular cells) → tubular injury → interstitial inflammation → fibrosis (TGF-β)
RAAS amplification: Angiotensin II is a key driver — it causes efferent arteriolar constriction (↑GFR pressure), promotes TGF-β and aldosterone release, and drives hypertension. This is why ACE inhibitors and ARBs slow CKD progression — they break the RAAS-driven intraglomerular hypertension loop and reduce proteinuria.
Examination point: The Brenner 'intact nephron hypothesis' (surviving nephrons compensate at a cost) explains why CKD is progressive and why reducing proteinuria and blood pressure are the two most evidence-based interventions to slow it.
Uraemia — Systemic Complications
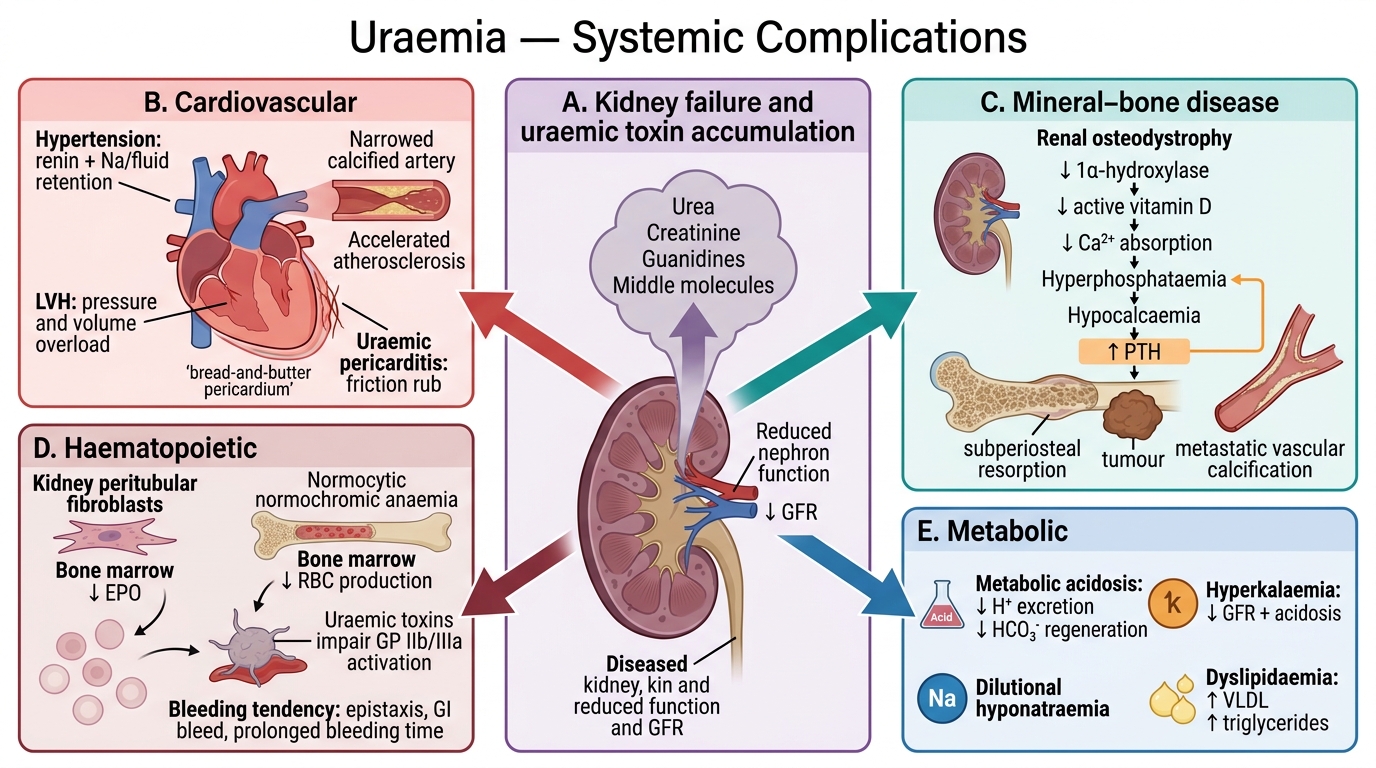
Systemic Complications of Uraemia
Uraemia is the clinical syndrome resulting from the accumulation of nitrogenous waste products (urea, creatinine, guanidines, 'middle molecules') and the metabolic consequences of kidney failure. It affects virtually every organ system.
Cardiovascular (leading cause of death in CKD):
- Hypertension — ↑renin, Na/fluid retention
- Left ventricular hypertrophy — pressure and volume overload
- Accelerated atherosclerosis — inflammation, dyslipidaemia, calcification
- Uraemic pericarditis — serofibrinous exudate; 'bread-and-butter' pericardium on gross examination; chest pain + friction rub
Renal Osteodystrophy / Mineral–Bone Disease:
- ↓Renal 1α-hydroxylase → ↓active vitamin D (1,25-dihydroxycholecalciferol)
- ↓Vitamin D → ↓intestinal Ca²⁺ absorption → hypocalcaemia
- ↓Renal phosphate excretion → hyperphosphataemia → further ↓iCa²⁺ (Ca²⁺-PO₄ binding)
- Hypocalcaemia + ↓vitamin D → ↑PTH (secondary hyperparathyroidism) → osteitis fibrosa cystica, Brown tumours, subperiosteal bone resorption
- Vascular calcification (metastatic)
Haematopoietic:
- Normocytic normochromic anaemia — ↓erythropoietin (EPO) production by peritubular fibroblasts → ↓RBC production
- Platelet dysfunction (uraemic toxins impair GP IIb/IIIa activation) → bleeding tendency (epistaxis, GI bleed, prolonged bleeding time)
Metabolic:
- Metabolic acidosis (↓H⁺ excretion, ↓HCO₃⁻ regeneration)
- Hyperkalaemia (↓GFR + acidosis)
- Hyponatraemia (dilutional)
- Dyslipidaemia (↑VLDL, ↑triglycerides)
Neurological:
- Uraemic encephalopathy — confusion, asterixis (flapping tremor), seizures, coma
- Peripheral neuropathy — glove-and-stocking sensorimotor; restless leg syndrome
Gastrointestinal:
- Nausea, vomiting, anorexia (uraemic gastritis)
- Uraemic frost (urea crystallises on skin) — late sign in severe uraemia
Immunological: Impaired neutrophil and lymphocyte function → ↑susceptibility to infection
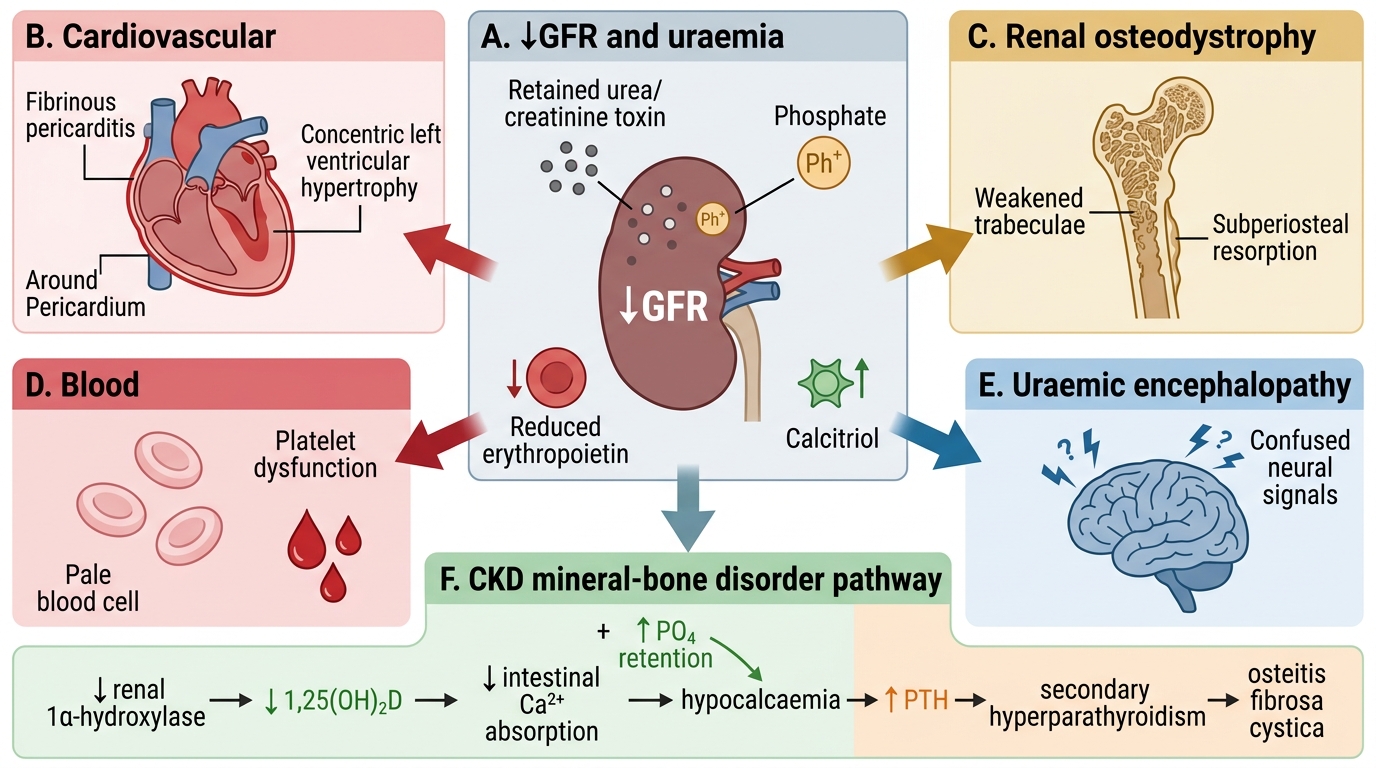
Systemic Complications of Uraemia in Chronic Kidney Disease
SELF-CHECK
A CKD Stage G4 patient has haemoglobin 7.8 g/dL (normocytic normochromic), serum calcium 7.2 mg/dL, phosphate 6.8 mg/dL, and PTH 420 pg/mL (normal <65). What is the sequence of events causing his elevated PTH?
A. ↑Phosphate retention → ↑PTH directly (independent of calcium)
B. ↓Renal 1α-hydroxylase → ↓1,25-OH₂D → ↓Ca²⁺ absorption + ↑PO₄ retention → hypocalcaemia → ↑PTH (secondary hyperparathyroidism)
C. ↑Vitamin D production by diseased kidney → hypercalcaemia → ↑PTH
D. ↓EPO → anaemia → bone marrow compensates by absorbing more calcium → hypocalcaemia → ↑PTH
Reveal Answer
Answer: B. ↓Renal 1α-hydroxylase → ↓1,25-OH₂D → ↓Ca²⁺ absorption + ↑PO₄ retention → hypocalcaemia → ↑PTH (secondary hyperparathyroidism)
The sequence is: damaged kidney → ↓1α-hydroxylase activity → ↓1,25(OH)₂D3 (active vitamin D) → ↓intestinal calcium absorption AND ↑phosphate retention (↓renal excretion) → phosphate binds serum calcium → hypocalcaemia → parathyroid glands stimulated → ↑PTH (secondary hyperparathyroidism). Prolonged PTH elevation causes osteitis fibrosa cystica. Correction requires active vitamin D analogues (calcitriol/alfacalcidol) + phosphate binders.