Page 8 of 25
PA27.5-6 | Glomerular Diseases — SDL Guide (Part 3)
Nephritic Syndrome: Overview and Pathophysiology
Nephritic Syndrome: Glomerular Inflammation and Diagnostic Clues
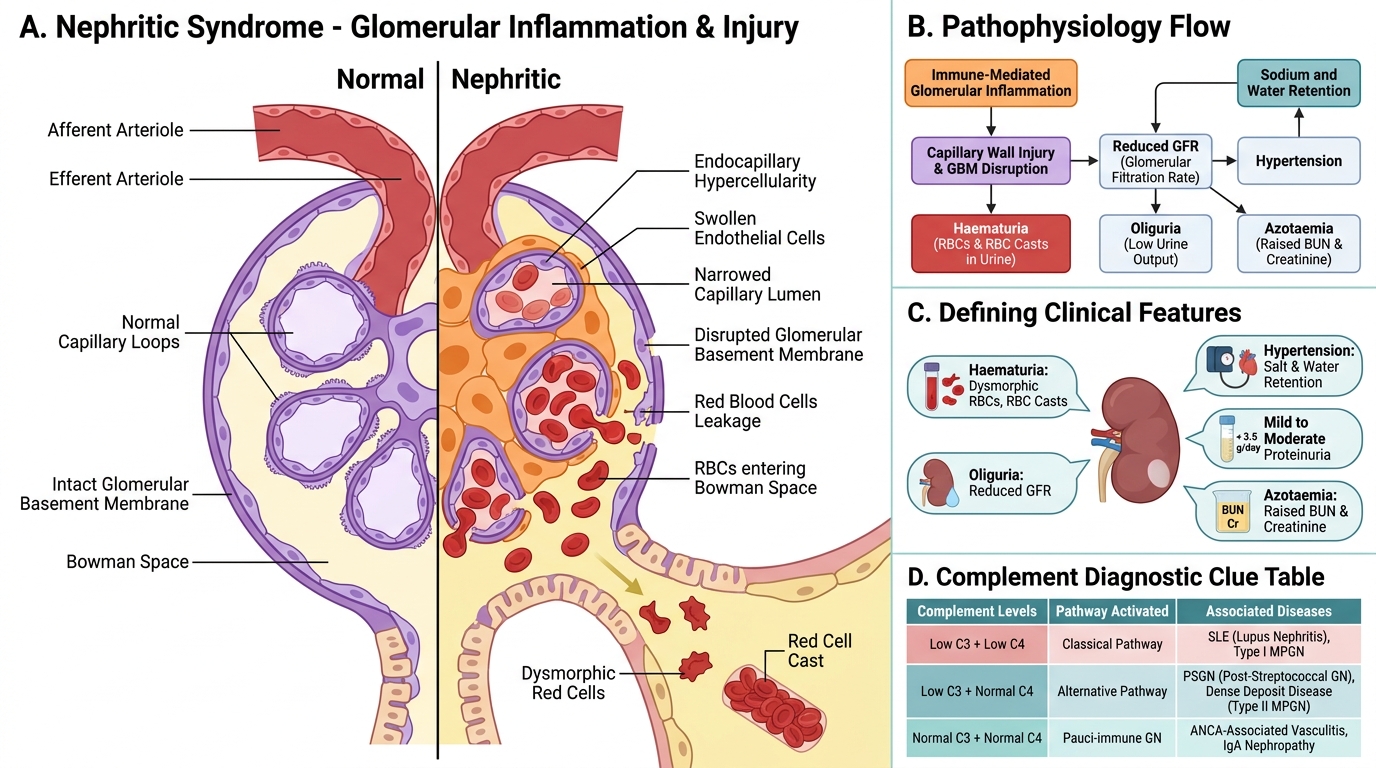
Nephritic syndrome reflects glomerular inflammation with disruption of the capillary wall, leading to haematuria and red cell leakage. Its five defining features are:
1. Haematuria — dysmorphic red cells and red cell casts in urine (the pathognomonic finding)
2. Oliguria — reduced GFR due to hypercellularity and capillary compression
3. Hypertension — sodium and water retention, renin release
4. Proteinuria — mild to moderate (< 3.5 g/day), not as heavy as nephrotic)
5. Azotaemia — elevated BUN and creatinine
Why haematuria? Inflammatory injury to the capillary wall creates gaps that allow red cells to squeeze through. As they traverse the tubules, they are moulded into red cell casts in the concentrated acidic tubular fluid. A single red cell cast on urine microscopy is sufficient to establish the diagnosis of glomerulonephritis.
Complement as a diagnostic clue:
• Low C3, low C4 → Classical pathway activation (SLE, Type I MPGN)
• Low C3, normal C4 → Alternative pathway (PSGN, Dense Deposit Disease, ANCA vasculitis — wait, ANCA is pauci-immune, see below)
• Normal C3 and C4 → Pauci-immune GN (ANCA-RPGN), IgA nephropathy
This complement profile guides the differential diagnosis even before biopsy.
Post-Streptococcal / Post-Infectious GN
Post-Streptococcal Glomerulonephritis: Pathogenesis and Pathology
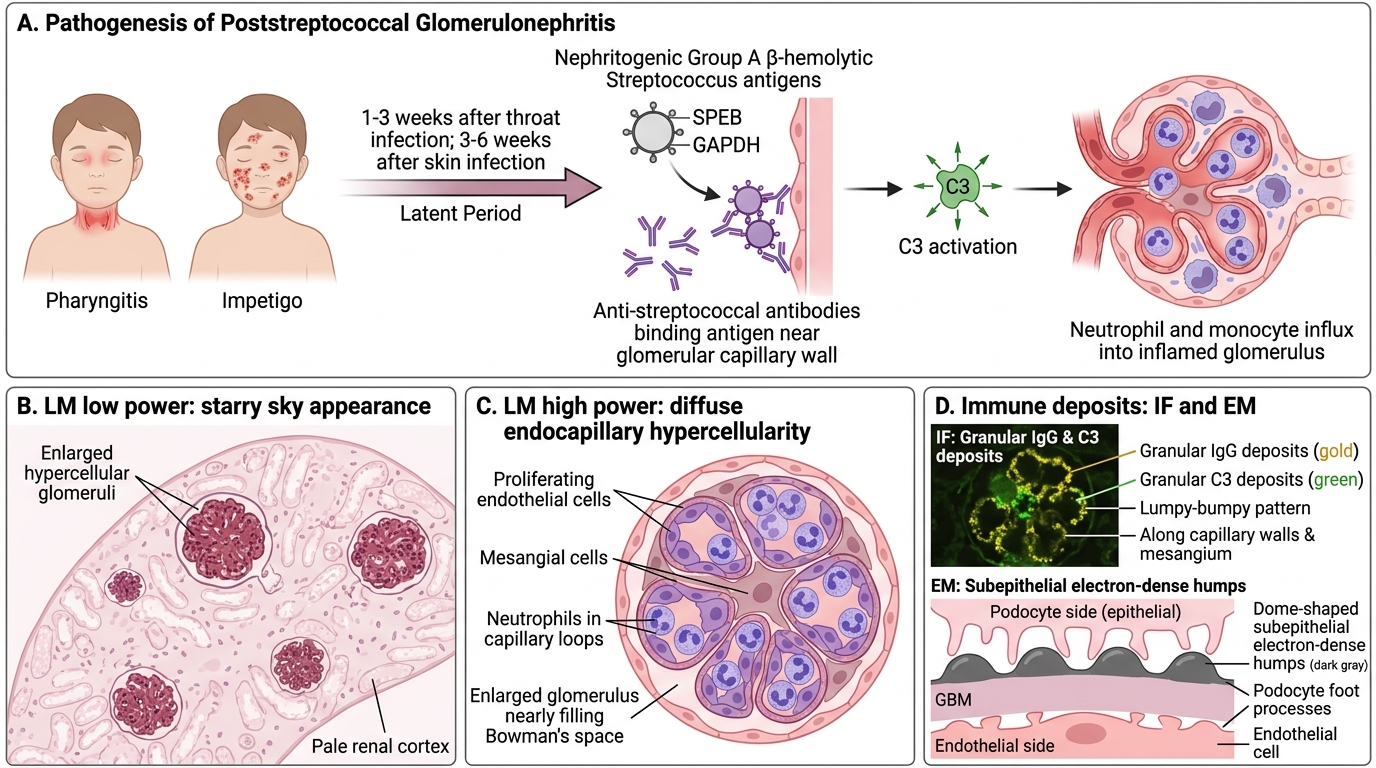
Post-streptococcal glomerulonephritis (PSGN) is the archetype of immune-complex–mediated nephritic syndrome, caused by nephritogenic strains of Group A β-haemolytic Streptococcus.
Timing: Latent period 1-3 weeks after throat infection (pharyngitis) or 3-6 weeks after skin infection (impetigo). The latency reflects time for immune-complex formation and deposition.
Who gets it? Children 5-12 years; males > females. Now rare in high-income countries due to antibiotic use; still common in India (skin infections, crowded conditions).
Pathogenesis: Nephritogenic streptococcal antigens (SPEB — cysteine protease; GAPDH) are deposited in subepithelial space → circulating anti-streptococcal antibodies arrive and bind in situ → immune complexes activate complement (C3) → neutrophil and monocyte influx → hypercellularity and injury.
Pathology:
• LM: Diffuse endocapillary hypercellularity (proliferating endothelial and mesangial cells + infiltrating neutrophils). The glomerulus is enlarged and hypercellular, filling Bowman's space. Described as the 'starry sky' appearance on low power (hypercellular glomeruli scattered in a pale background).
• IF: Granular IgG and C3 deposits in a 'lumpy-bumpy' pattern on the capillary walls and mesangium.
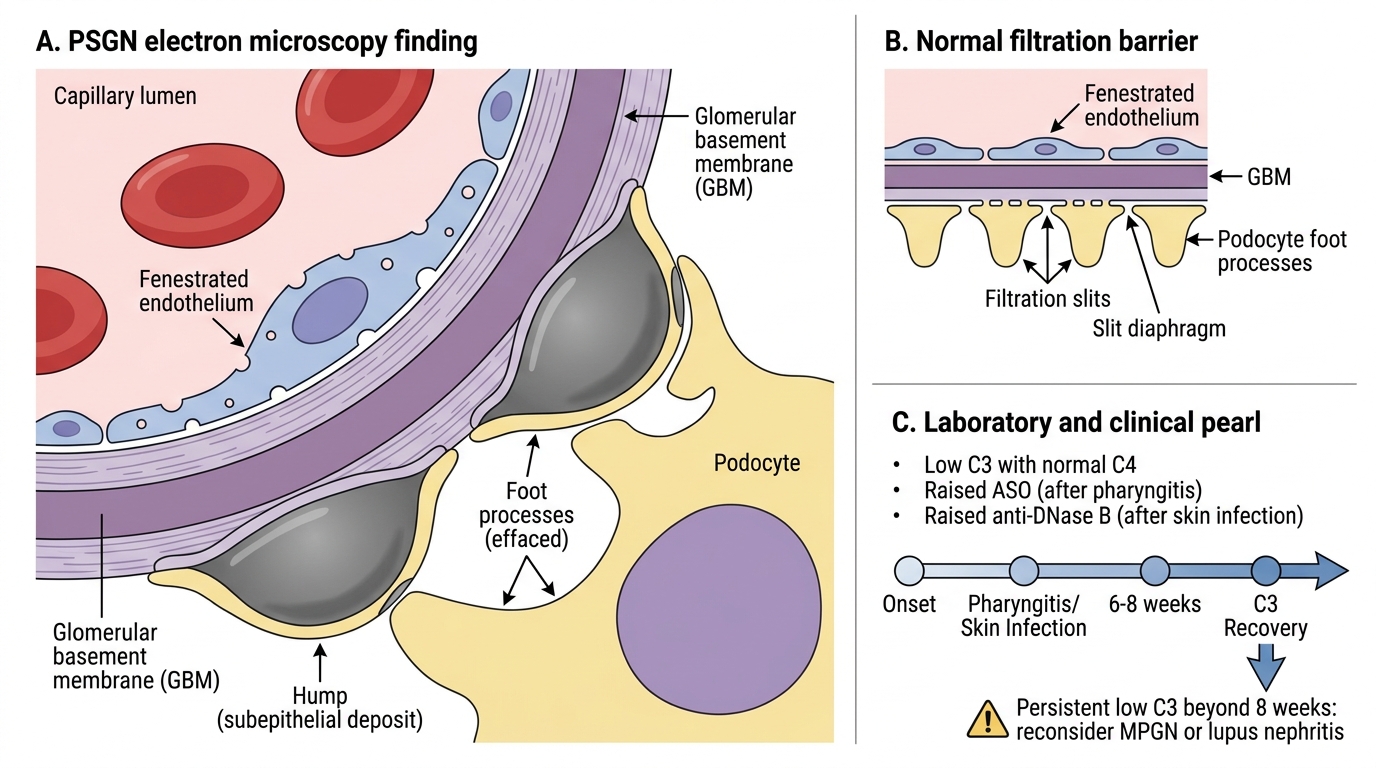
• EM: Subepithelial electron-dense deposits in a hump-shaped (dome-shaped) configuration — the pathognomonic 'subepithelial humps' of PSGN. These humps sit on the epithelial side of the GBM like camel humps.
Post-Streptococcal GN: Subepithelial Humps on EM
Laboratory: Low C3 (alternative pathway consumption), normal C4 in most. Anti-streptolysin O (ASO) titre raised after pharyngitis; anti-DNase B after skin infection. Elevated.
Clinical course: Self-limiting in children — 95%+ full recovery. Elderly patients and those with pre-existing kidney disease have a worse prognosis. Steroids not indicated. Treat the streptococcal infection with penicillin (to stop spread, not to alter renal course).
CLINICAL PEARL
In PSGN the complement drops early and recovers within 6-8 weeks. If C3 remains low beyond 8 weeks, reconsider the diagnosis — think MPGN (persistent low complement) or lupus nephritis (both C3 and C4 low). This 'complement recovery rule' is a high-yield clinical pearl used in differential diagnosis of low-complement nephritic syndrome.
Rapidly Progressive GN (RPGN) / Crescentic GN
Rapidly Progressive Crescentic Glomerulonephritis
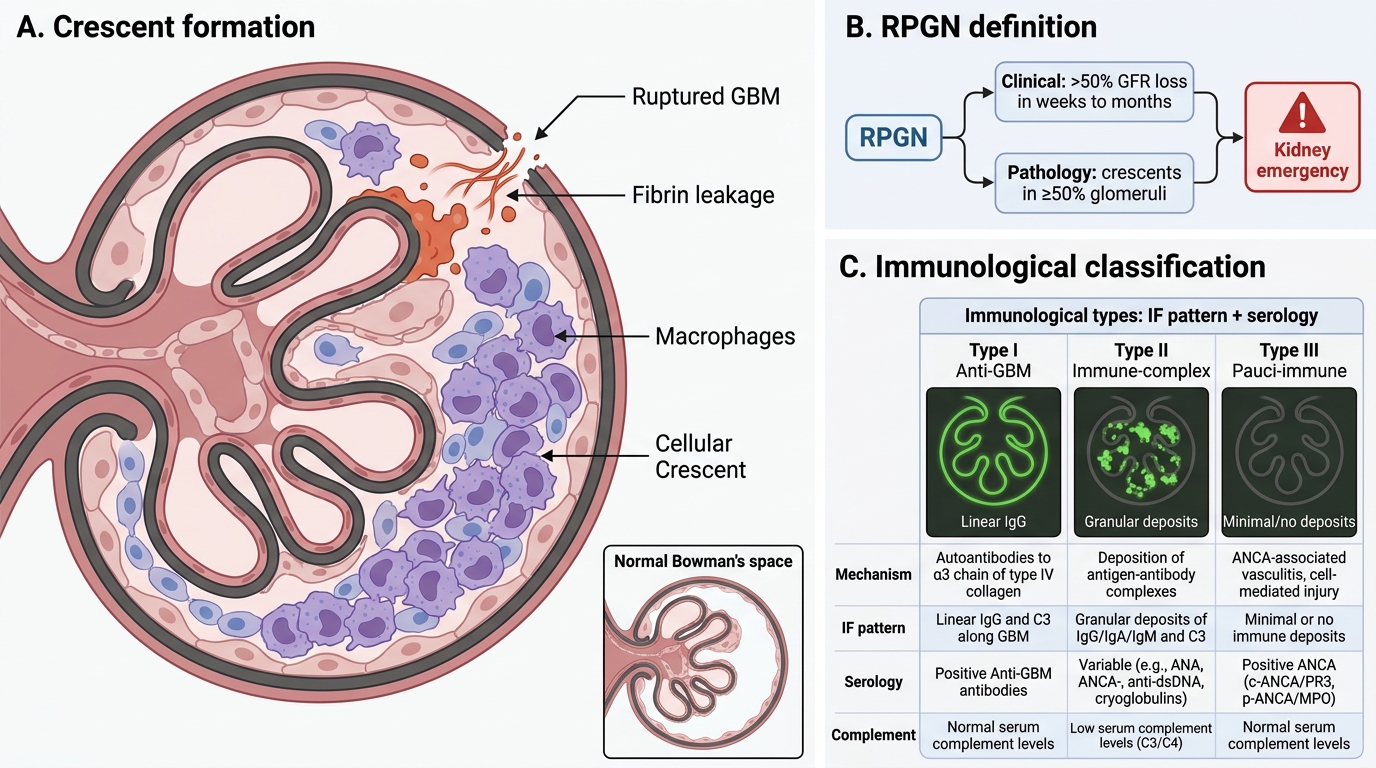
Rapidly progressive glomerulonephritis (RPGN) is defined clinically as a loss of > 50% GFR within weeks to months, and pathologically by the presence of crescents in ≥ 50% of glomeruli.
What is a crescent? Fibrin and proliferating parietal epithelial cells of Bowman's capsule fill Bowman's space in a crescent shape in response to fibrin leakage through ruptured GBM. Macrophages also contribute. Crescents are a morphological marker of severe, destructive glomerular injury — a kidney emergency.
Three immunological types — memorise IF + serology:
| Type | Mechanism | IF Pattern | Serology | Complement |
|---|---|---|---|---|
| Type I (Anti-GBM) | Anti-GBM antibodies (Goodpasture) | Linear IgG | Anti-GBM Ab (+) | Normal or mildly ↓ |
| Type II (Immune-complex) | IC deposition (post-infectious, lupus, IgA) | Granular IgG/C3 | Variable | Low |
| Type III (Pauci-immune) | ANCA vasculitis (MPA, GPA) | Negative (pauci = few/none) | ANCA (+): MPO (p-ANCA) or PR3 (c-ANCA) | Normal |
Type III is the most common (~60%) and is associated with ANCA-associated small vessel vasculitis — microscopic polyangiitis (MPO/p-ANCA) or granulomatosis with polyangiitis/Wegener's (PR3/c-ANCA).
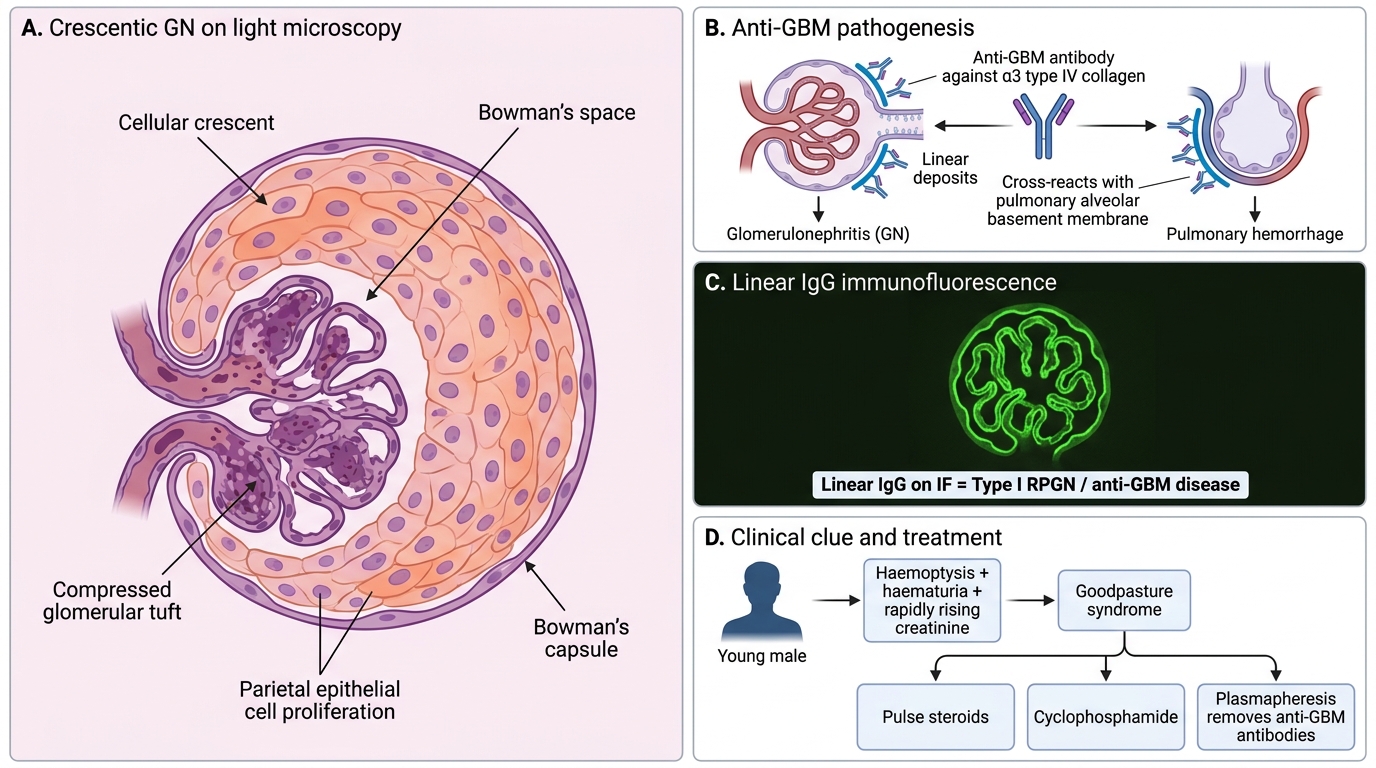
Crescentic GN and Goodpasture Syndrome
Goodpasture syndrome (Type I): Anti-GBM antibodies cross-react with pulmonary alveolar basement membrane (same α3 type IV collagen) → pulmonary haemorrhage + RPGN. HLA-DR15 predisposition. Young males. Haemoptysis + haematuria = Goodpasture until proven otherwise.
Treatment principle: All RPGN types require aggressive immunosuppression (pulse steroids + cyclophosphamide). Type I adds plasmapheresis to remove anti-GBM antibodies. Without treatment, ESRD within weeks.
SELF-CHECK
A 25-year-old man presents with haemoptysis and rapidly worsening renal function. Renal biopsy shows crescents in 70% of glomeruli with LINEAR IgG on immunofluorescence. Which serology is most likely positive?
A. Anti-neutrophil cytoplasmic antibodies (ANCA/MPO)
B. Anti-GBM antibodies
C. Anti-dsDNA antibodies
D. Anti-streptolysin O titre
Reveal Answer
Answer: B. Anti-GBM antibodies
Linear IgG on IF is the hallmark of anti-GBM antibody disease (Type I RPGN). The combination of pulmonary haemorrhage and rapidly progressive GN in a young male with linear IgG is classic Goodpasture syndrome — caused by autoantibodies against the NC1 domain of the α3 chain of type IV collagen. ANCA is associated with pauci-immune (Type III) RPGN, which shows a NEGATIVE IF.