Page 14 of 27
PA27.{14,16} | Renal & Urothelial Tumours — SDL Guide (Part 2)
RCC — Gross Pathology and Staging
Renal Cell Carcinoma: Gross Pathology and Staging
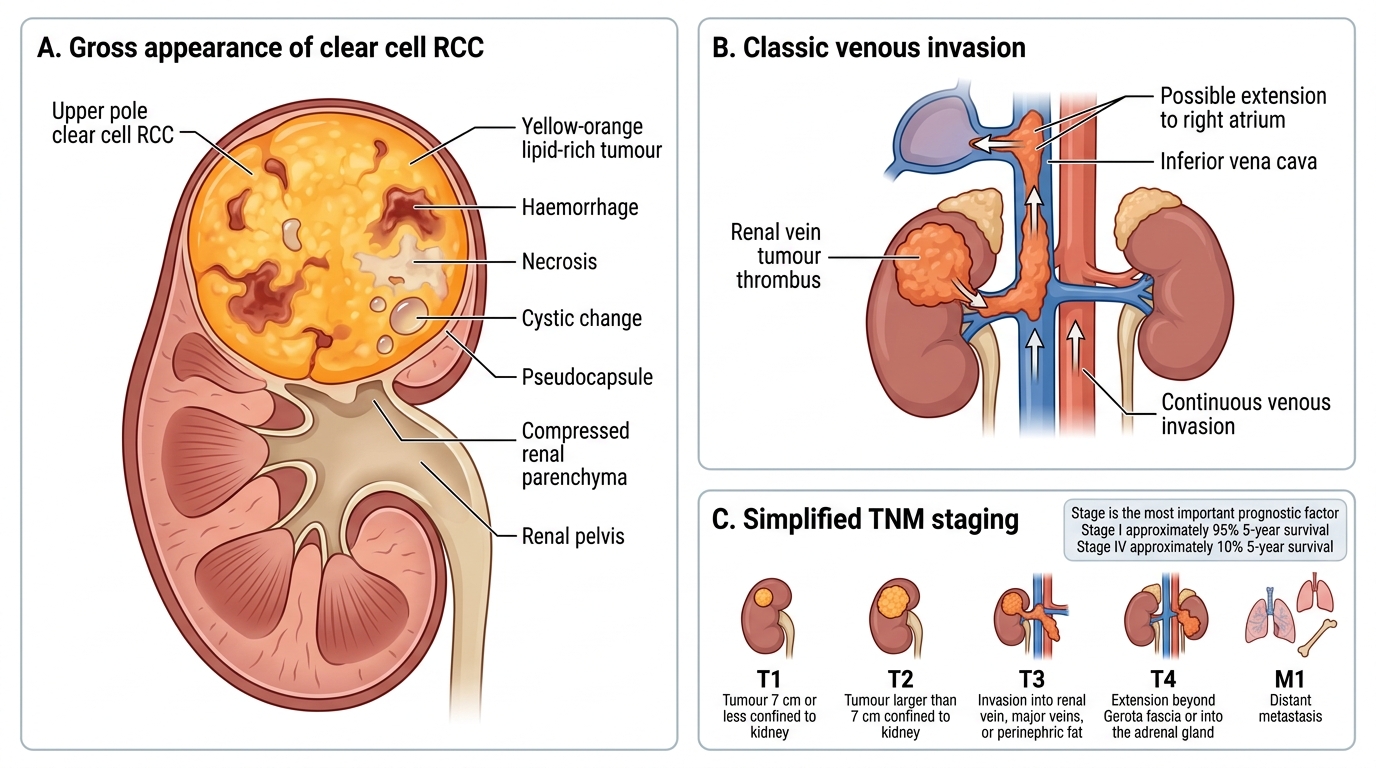
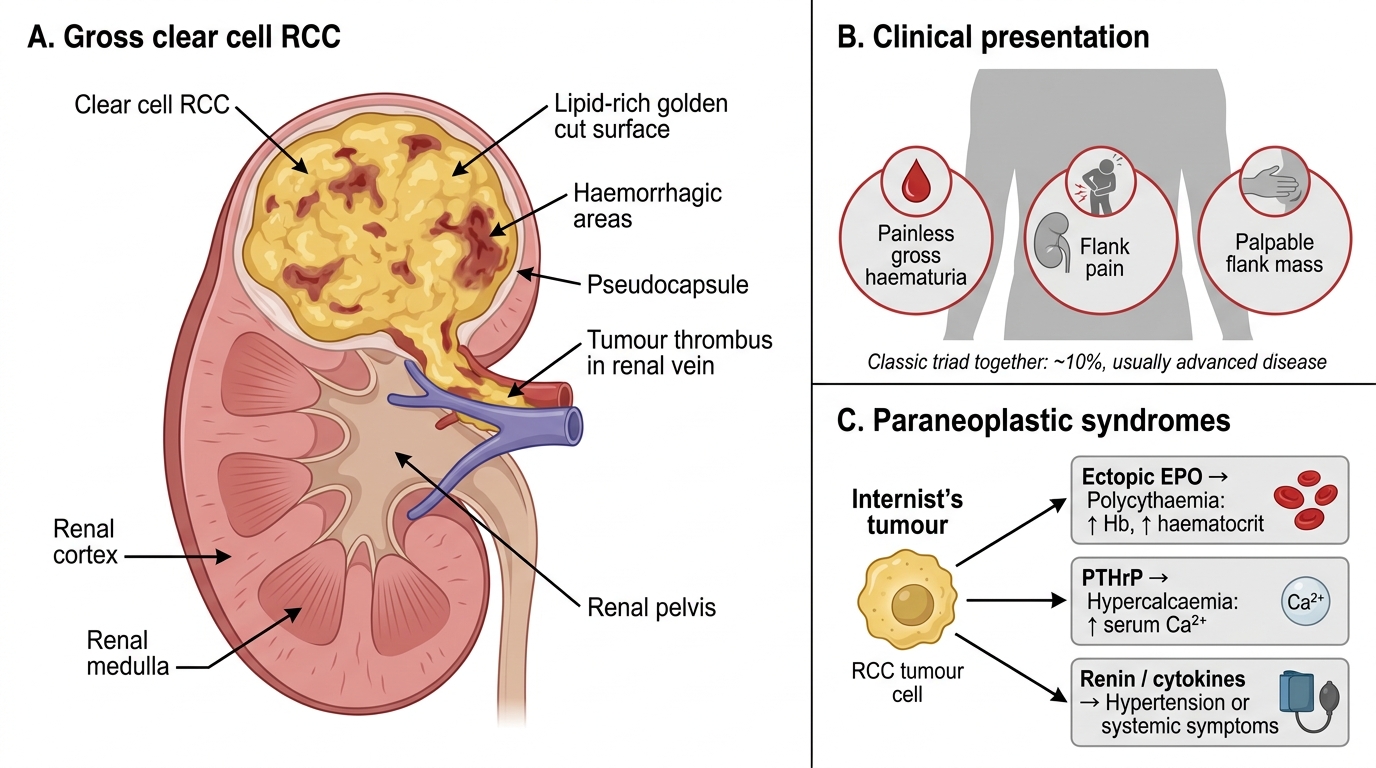
Gross features of clear cell RCC (the prototype):
- Location: upper pole preference (70%)
- Appearance: spherical mass, bright yellow-orange cut surface due to lipid content; haemorrhage and necrosis create a variegated pattern; cystic change common
- Pseudocapsule: often present, giving a false sense of containment
- Venous invasion: a critical and classic feature — RCC extends as a continuous tumour thrombus up the renal vein and into the inferior vena cava (IVC); the thrombus can reach the right atrium
Staging (simplified TNM for examination):
| Stage | Criteria |
|---|---|
| T1 | ≤7 cm, confined to kidney |
| T2 | >7 cm, confined to kidney |
| T3 | Into major veins / perinephric fat (not beyond Gerota fascia) |
| T4 | Beyond Gerota fascia / into adrenal |
| M1 | Distant metastasis |
Stage is the single most important prognostic factor. 5-year survival: Stage I ~95%, Stage IV ~10%.
Clear Cell Renal Cell Carcinoma: Gross Pathology and Clinical Correlates
RCC — Clinical Features and Paraneoplastic Syndromes
Renal Cell Carcinoma: Clinical Features and Paraneoplastic Syndromes
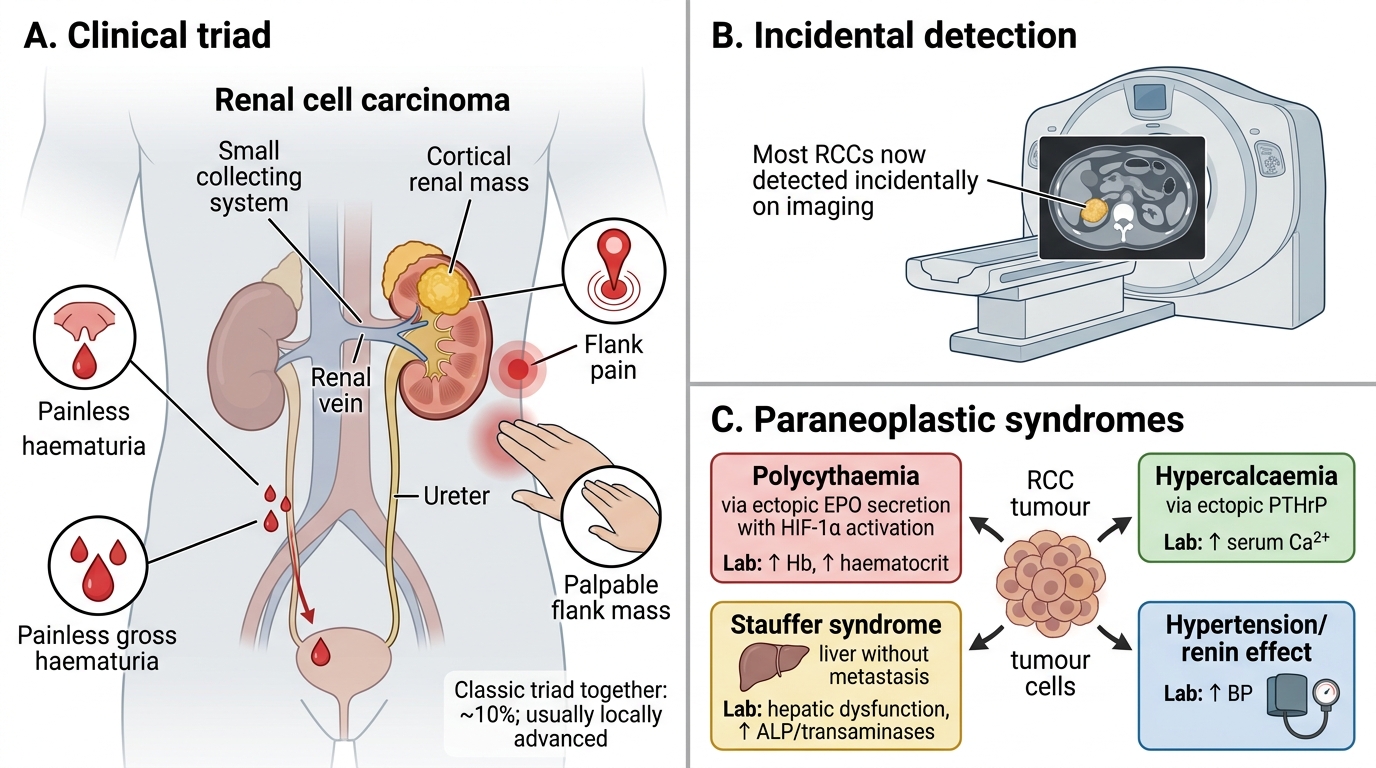
Classic triad:
- Haematuria (most common single symptom — painless, gross)
- Flank pain
- Palpable flank mass
Importantly, all three together (the 'classic triad') is present in only ~10% of cases and usually indicates locally advanced disease. Most RCCs are now found incidentally on imaging done for other reasons.
Paraneoplastic syndromes (high-yield): RCC has more paraneoplastic associations than almost any other solid tumour — nicknamed the 'internist's tumour'.
| Syndrome | Mechanism | Lab finding |
|---|---|---|
| Polycythaemia | Ectopic EPO secretion by tumour cells (HIF-1α → EPO gene) | ↑ Hb, ↑ haematocrit |
| Hypercalcaemia | Ectopic PTHrP secretion | ↑ Serum Ca²⁺ |
| Stauffer syndrome | Non-metastatic hepatic dysfunction (mechanism unclear, possibly cytokine-mediated) | ↑ LFTs without liver mets |
| Hypertension | Ectopic renin; vascular compression | Refractory HTN |
| Cushing syndrome | Ectopic ACTH (rare) | Hypercortisolaemia |
Remember that resolution of paraneoplastic syndromes after nephrectomy is evidence of their tumour origin.
Spread:
• Haematogenous — most common route; characteristic 'cannonball' metastases in the lungs (multiple, well-defined round nodules); also bone, brain, liver, opposite kidney
• Lymphatic — para-aortic and paracaval nodes
• Direct — adrenal gland, perinephric fat, Gerota fascia
• Notorious for late metastasis — may present 10–20 years after nephrectomy ('late recurrence')
• Venous invasion and tumour thrombus (as above)
SELF-CHECK
A 55-year-old woman is found to have a renal mass incidentally. Histology shows nests of cells with optically clear cytoplasm separated by thin-walled sinusoidal vessels. Genetic analysis shows deletion at chromosome 3p25. Which protein is most directly responsible for the upregulated angiogenesis in this tumour?
A. p53
B. HIF-1α
C. BRCA1
D. c-MYC
Reveal Answer
Answer: B. HIF-1α
Loss of VHL (chromosome 3p25) prevents HIF-1α from being targeted for proteasomal degradation. Accumulated HIF-1α transcriptionally upregulates VEGF, PDGF, and EPO — driving angiogenesis, tumour growth, and paraneoplastic polycythaemia. p53, BRCA1, and c-MYC are not the primary drivers in clear cell RCC.
CLINICAL PEARL
The 'internist's tumour' rule: Whenever a clinical scenario gives you an unexplained polycythaemia, hypercalcaemia, or non-metastatic hepatic dysfunction (Stauffer syndrome) in a middle-aged man with a flank mass or haematuria, think RCC first. These paraneoplastic syndromes arise because HIF-1α drives ectopic EPO secretion, while tumour-derived PTHrP causes hypercalcaemia. Crucially, all three may resolve after nephrectomy — a point that often appears in MCQs.
Another high-yield point: 'cannonball' lung metastases on a chest X-ray in a patient with a renal mass = RCC until proven otherwise. The well-defined, multiple round nodules reflect haematogenous spread via the renal vein and IVC.
Wilms Tumour (Nephroblastoma)
Wilms Tumour: Gross, Genetics and Triphasic Histology
Wilms tumour is the commonest renal tumour of childhood, typically presenting between ages 2–5 years. It is one of the most successfully treated paediatric solid tumours, with overall survival >90% with modern multimodal therapy.
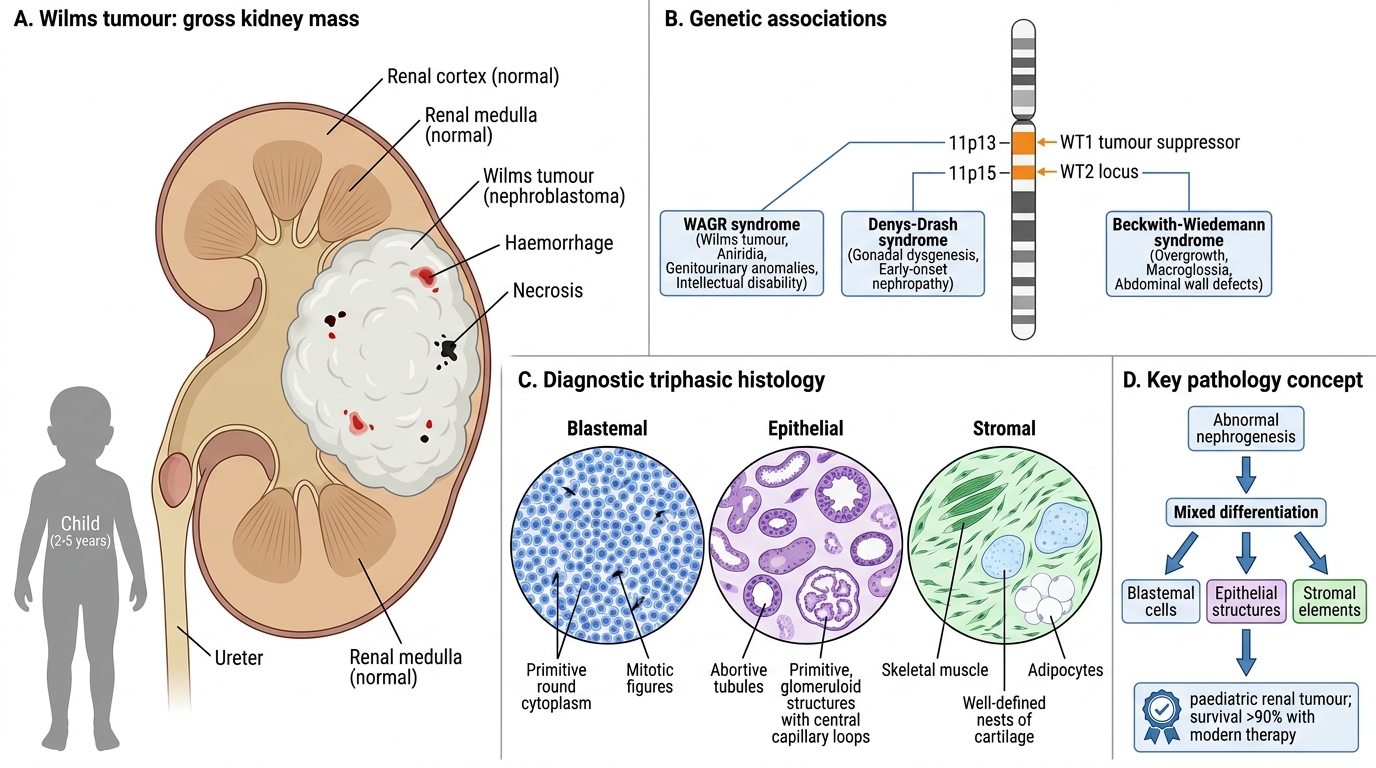
Genetics:
• WT1 gene (chromosome 11p13) — tumour suppressor encoding a transcription factor essential for normal kidney development; loss → failure of terminal differentiation
• WT2 locus (11p15) — associated with Beckwith–Wiedemann syndrome (macroglossia, hemihypertrophy, organomegaly + Wilms)
• WAGR syndrome: Wilms, Aniridia, Genitourinary anomalies, (intellectual) Retardation — 11p13 deletion
• Denys–Drash syndrome: WT1 point mutation → Wilms + diffuse mesangial sclerosis + male pseudohermaphroditism
Pathology — the diagnostic triphasic histology:
Wilms tumour recapitulates abnormal nephrogenesis. The classic pattern is triphasic:
1. Blastemal component — small, round, densely packed primitive cells with scant cytoplasm (resembles the metanephric blastema); the most cellular, mitotically active component
2. Epithelial component — abortive tubular and glomerular structures; recapitulates nephron formation
3. Stromal component — loose mesenchyme, may contain skeletal muscle, cartilage, or fat
Not all three components need be present in every tumour, but the triphasic pattern is the classic diagnostic image.
Gross: Large, well-circumscribed, soft, fish-flesh grey-white tumour, often with areas of haemorrhage, necrosis, and cyst formation. Pseudocapsule is common. Usually unilateral (5% bilateral — associated with WT1 germline mutations).
Clinical presentation:
• Painless abdominal mass in a child — the classic and most common presentation; often noticed by the parent while bathing
• Abdominal pain, haematuria, hypertension (less common)
• Never cross the midline on palpation (unlike neuroblastoma, which often does)
Unfavourable histology: Presence of anaplasia (diffuse > focal) is the strongest adverse prognostic factor.
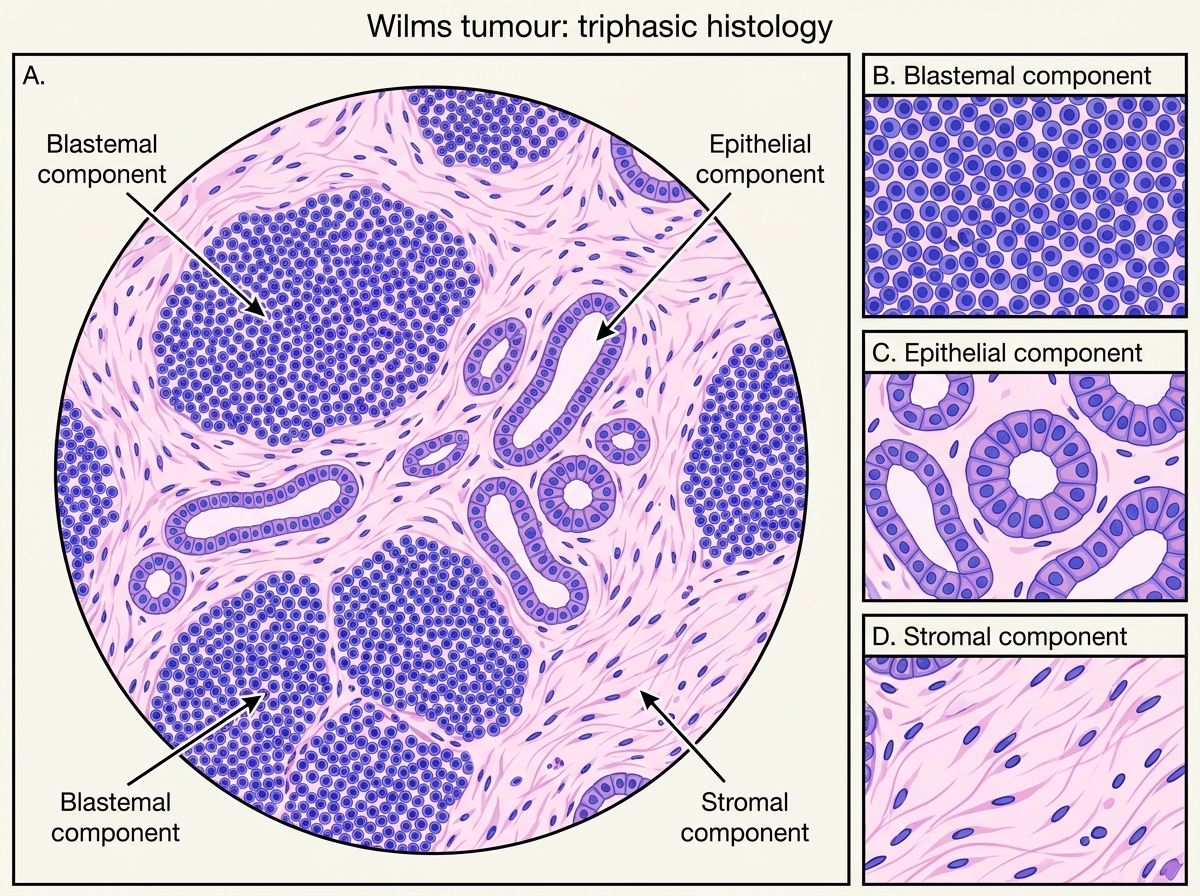
Wilms Tumour: Triphasic Histology
SELF-CHECK
A 3-year-old child presents with a painless abdominal mass that does not cross the midline. CT shows a large unilateral renal mass. Histology demonstrates primitive tubular structures, dense small blue cells, and loose mesenchyme. Which gene mutation is most likely responsible?
A. RB1 on chromosome 13q14
B. WT1 on chromosome 11p13
C. VHL on chromosome 3p25
D. MYCN on chromosome 2p24
Reveal Answer
Answer: B. WT1 on chromosome 11p13
The triphasic histology (blastemal + epithelial + stromal) is pathognomonic of Wilms tumour (nephroblastoma). WT1 (chromosome 11p13) is the classic gene implicated. RB1 loss causes retinoblastoma; VHL loss causes clear cell RCC; MYCN amplification is characteristic of neuroblastoma — which can also present as an abdominal mass but crosses the midline and has adrenal origin.