Page 15 of 32
PA25.5 | Occupational & Interstitial Lung Disease — SDL Guide (Part 3)
Hypersensitivity Pneumonitis
Hypersensitivity Pneumonitis: Antigens, Mechanism, and Morphology
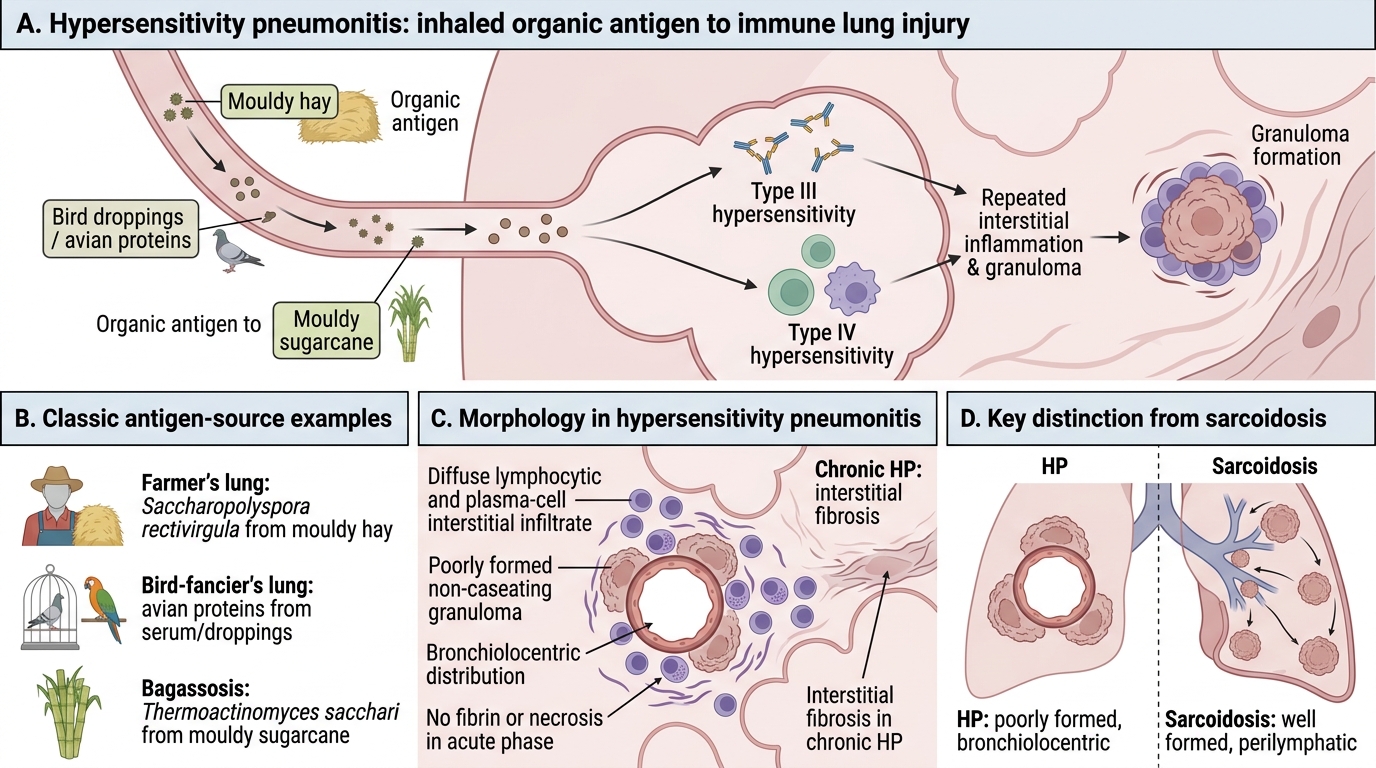
Hypersensitivity pneumonitis (HP), also called extrinsic allergic alveolitis, is an immunologically mediated ILD caused by repeated inhalation of organic antigens — in contrast to the mineral dust diseases above.
Mechanism: Combined Type III (immune complex) + Type IV (cell-mediated, granulomatous) hypersensitivity.
Classic examples:
| Syndrome | Antigen | Source |
|---|---|---|
| Farmer's lung | Saccharopolyspora rectivirgula (thermophilic actinomycete) | Mouldy hay |
| Bird-fancier's lung | Avian proteins (serum, droppings) | Pigeons, parrots |
| Bagassosis | Thermoactinomyces sacchari | Mouldy sugarcane |
Morphology: Diffuse lymphocytic and plasma-cell interstitial infiltrate; poorly formed non-caseating granulomas (bronchiolocentric); no fibrin or necrosis in acute phase. Chronic HP → interstitial fibrosis.
Key distinction from sarcoidosis: HP granulomas are poorly-formed and bronchiolocentric; sarcoidosis granulomas are well-formed and perilymphatic. HP has a clear antigen exposure history; sarcoidosis does not.
Outcome: Removal from antigen exposure → resolution in acute/subacute HP. Chronic continued exposure → irreversible fibrosis.
Idiopathic Pulmonary Fibrosis (UIP Pattern)
Idiopathic Pulmonary Fibrosis: UIP Pattern
Idiopathic pulmonary fibrosis (IPF) is the most common and most lethal idiopathic ILD. It has no known exogenous cause.
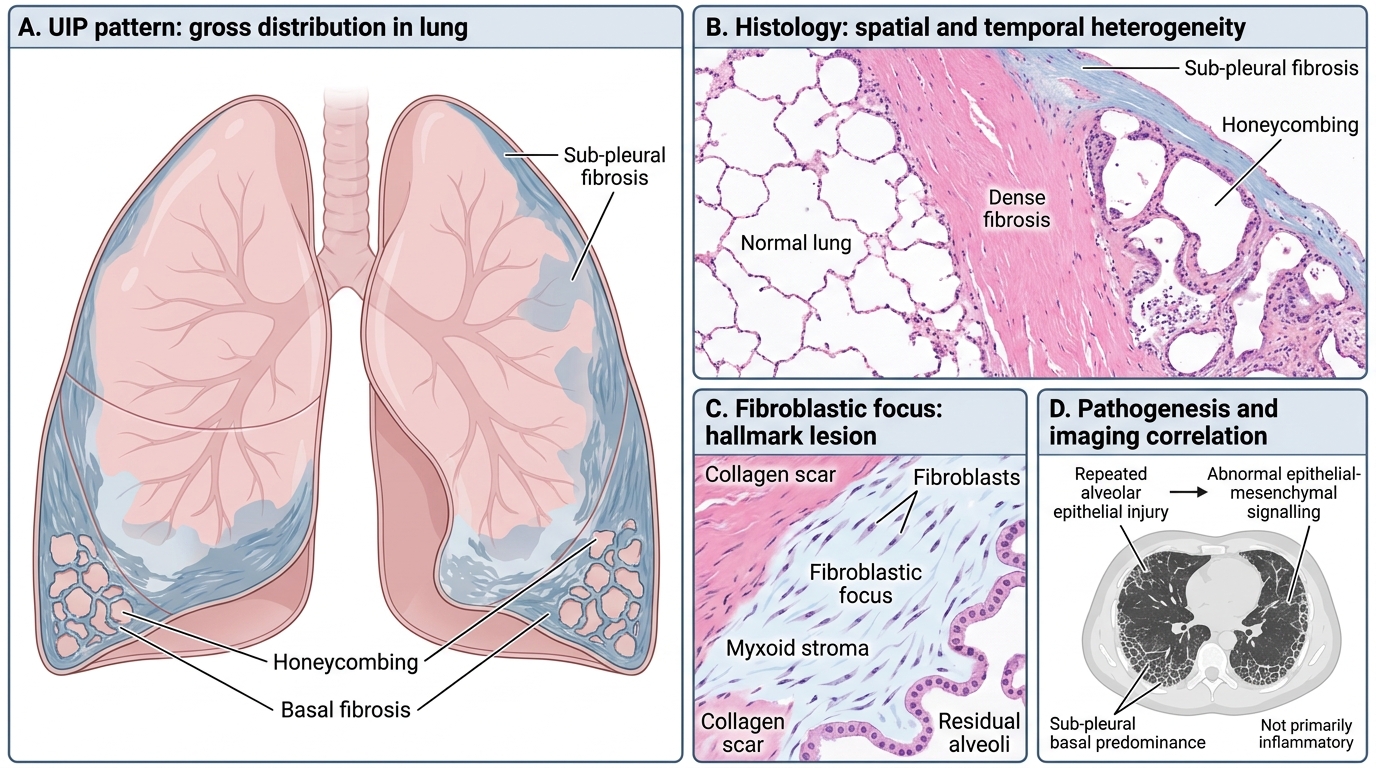
Histological pattern: Usual Interstitial Pneumonia (UIP)
• Spatial and temporal heterogeneity — areas of normal lung adjacent to fibrosis and honeycombing (temporal heterogeneity)
• Fibroblastic foci — the hallmark: clusters of actively proliferating fibroblasts in a myxoid stroma at the advancing edge of fibrosis
• Sub-pleural, basal predominance
• Honeycombing (cystic spaces lined by bronchiolar epithelium) in advanced disease
High-resolution CT (HRCT): Bilateral, sub-pleural, basal-predominant reticular opacities ± honeycombing — the UIP pattern on imaging.
Pathogenesis: Current model — repeated alveolar epithelial injury (from micro-aspirations, smoking, viral infections?) → abnormal epithelial-mesenchymal signalling → fibroblast activation that fails to switch off. NOT primarily inflammatory (anti-inflammatory drugs ineffective).
Prognosis: Median survival 2–3 years from diagnosis. Anti-fibrotic agents (pirfenidone, nintedanib) slow progression but do not reverse fibrosis.
Sarcoidosis
Sarcoidosis: Granulomas, Inclusions, and Lung Staging
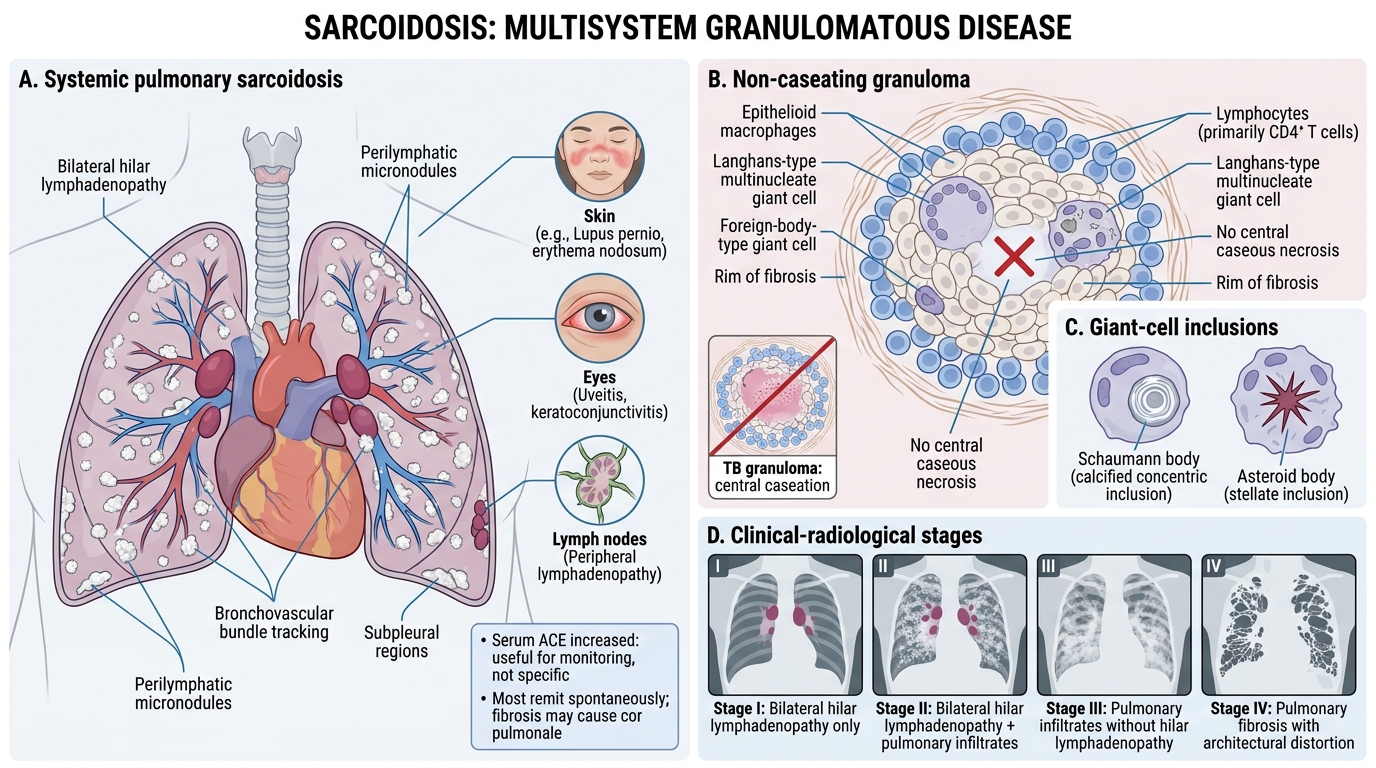
Sarcoidosis is a systemic granulomatous disease of unknown aetiology affecting lungs (90%), lymph nodes, skin, eyes, and other organs.
Key pathological feature: non-caseating granulomas
• Well-formed, discrete aggregates of epithelioid macrophages (activated) with multinucleate giant cells (Langhans and foreign-body type)
• No central necrosis (caseation) — distinguishes from TB
• Surrounded by lymphocytes; outer rim of fibrosis in older lesions
• Perilymphatic distribution (along bronchovascular bundles and sub-pleural)
Inclusions in giant cells:
• Schaumann bodies (calcified concentric laminar structures)
• Asteroid bodies (stellate inclusions)
Clinical-radiological stages (lungs):
• Stage I: Bilateral hilar lymphadenopathy (BHL) alone
• Stage II: BHL + pulmonary infiltrates
• Stage III: Pulmonary infiltrates, no BHL
• Stage IV: Pulmonary fibrosis
Serology: ↑ Serum ACE (angiotensin-converting enzyme) — not specific but useful for monitoring.
Outcome: Most patients (60–70%) remit spontaneously. ~20% develop chronic disease. End-stage fibrosis → cor pulmonale.
SELF-CHECK
A 35-year-old pigeon-keeper presents with progressive breathlessness, low-grade fever, and bilateral interstitial opacities. Lung biopsy shows poorly-formed bronchiolocentric granulomas with lymphocytic infiltrate and no caseation. Which diagnosis fits best?
A. Sarcoidosis
B. Miliary tuberculosis
C. Hypersensitivity pneumonitis (bird-fancier's lung)
D. Idiopathic pulmonary fibrosis
Reveal Answer
Answer: C. Hypersensitivity pneumonitis (bird-fancier's lung)
Bird-fancier's lung (hypersensitivity pneumonitis from avian proteins) produces poorly-formed, bronchiolocentric granulomas — in contrast to sarcoidosis (well-formed, perilymphatic) and TB (caseating). The pigeon-keeping history, the organic antigen exposure, and the bronchiolocentric pattern clinch the diagnosis. IPF shows fibroblastic foci and honeycombing, not granulomas. Miliary TB has numerous caseating granulomas.